Cиндром Пейтца-Йегерса
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Синдром Пейтца–Егерса (СПЕ) является редким наследственным заболеванием (генодерматозом) с аутосомно-доминантным типом наследования, обусловленным мутациями в гене STK11. В статье описан алгоритм диагностики СПЕ у детей на примере клинического случая. У пациентки 5 лет с характерной для СПЕ пигментацией слизистой в области губ, не имеющей отягощенного семейного анамнеза, проведено генетическое тестирование и выявлена мутация de novo в гене STK11 (c.543C>G, p.N181K). С учетом возможного полипоза при СПЕ выполнено эндоскопическое обследование желудочно-кишечного тракта, при котором обнаружены полипы желудка, в том числе множественные гамартомные полипы, что полностью соответствует клиническим критериям заболевания.
Представленный клинический пример свидетельствует о необходимости коллегиального наблюдения больных с СПЕ с привлечением дерматолога, генетика, врача эндоскопической диагностики, детского онколога (в том числе в связи с высоким риском развития злокачественных новообразований по мере взросления ребенка).
Ключевые слова
Об авторах
Т.С. Белышева: д.м.н., ведущий научный сотрудник научно-консультативного отделения НИИ детской онкологии и гематологии
115478, Москва, Каширское шоссе, 23
ФГБУН «Институт молекулярной биологии имени В.А. Энгельгардта Российской академии наук»; ФГБУ «НМИЦ ДГОИ им. Дмитрия Рогачева» Минздрава России
Россия
Т.В. Наседкина: д.б.н., профессор, ведущий научный сотрудник лаборатории биологических микрочипов ИМБ им. В.А. Энгельгардта РАН; ведущий научный сотрудник НМИЦ ДГОИ им. Дмитрия Рогачева
SPIN-код: 3741-8214
ГСП-1, 119991, Москва, ул. Вавилова, 32
117198, Москва, ул. Саморы Машела, 1
Т.Т. Валиев: д.м.н., заведующий отделением детской онкологии и гематологии (химиотерапия гемобластозов) № 1 НИИ детской онкологии и гематологии НМИЦ онкологии им. Н.Н. Блохина; профессор кафедры детской онкологии им. акад. Л.А. Дурнова РМАНПО
115478, Москва, Каширское шоссе, 23
125993, Москва, ул. Баррикадная, 2/1, стр. 1
Н.В. Матинян: д.м.н., заведующая отделением анестезиологии-реанимации и интенсивной терапии НИИ детской онкологии и гематологии НМИЦ онкологии им. Н.Н. Блохина
SPIN-код: 9829-6657
115478, Москва, Каширское шоссе, 23
О.А. Малихова: д.м.н., заведующая отделом эндоскопии НИИ клинической онкологии им. Н.Н. Трапезникова НМИЦ онкологии им. Н.Н. Блохина, профессор кафедры онкологии и паллиативной медицины РМАНПО
115478, Москва, Каширское шоссе, 23
125993, Москва, ул. Баррикадная, 2/1, стр. 1
ФГБУ «НМИЦ онкологии им. Н.Н. Блохина» Минздрава России; ФГБУН «Институт молекулярной биологии имени В.А. Энгельгардта Российской академии наук»; ФГБУ «НМИЦ ДГОИ им. Дмитрия Рогачева» Минздрава России
Россия
В.В. Семенова: врач-генетик НИИ детской онкологии и гематологии НМИЦ онкологии им. Н.Н. Блохина; аспирант лаборатории биологических микрочипов ИМБ им. В.А. Энгельгардта РАН; врач-генетик НМИЦ ДГОИ им. Дмитрия Рогачева
SPIN-код: 9014-2847
115478, Москва, Каширское шоссе, 23ГСП-1
119991, Москва, ул. Вавилова, 32
117198, Москва, ул. Саморы Машела, 1
В.М. Козлова: врач-генетик научно-консультативного отделения НИИ детской онкологии и гематологии
115478, Москва, Каширское шоссе, 23
Т.П. Казубская: старший научный сотрудник лаборатории клинической цитологии отдела морфологической и молекулярно-генетической диагностики опухолей НИИ клинической онкологии им. Н.Н. Трапезникова
SPIN-код: 5224-5820
115478, Москва, Каширское шоссе, 23
Я.В. Вишневская: к.м.н., врач патологоанатомического отделения НИИ клинической онкологии
115478, Москва, Каширское шоссе, 23
С.Н. Михайлова: к.м.н., заведующая научно-консультативным отделением НИИ детской онкологии и гематологии
115478, Москва, Каширское шоссе, 23
С.Р. Варфоломеева: д.м.н., профессор, директор НИИ детской онкологии и гематологии
115478, Москва, Каширское шоссе, 23
Список литературы
1. Schreibman I.R., Baker M., Amos C., McGarrity T.J. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100(2):476–90. doi:10.1111/j.1572-0241.2005.40237.x. PMID:15667510.
2. Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Höglund P., Järvinen H., Kristo P., Pelin K., Ridanpää M., Salovaara R., Toro T., Bodmer W., Olschwang S., Olsen A.S., Stratton M.R., de la Chapelle A., Aaltonen L.A. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature. 1998;391(6663):184–7. doi:10.1038/34432. PMID:9428765.
3. Altamish M., Dahiya R., Singh A.K., Mishra A., Aljabali A.A.A., Satija S., Mehta M., Dureja H., Prasher P., Negi P., Kapoor D.N., Goyal R., Tambuwala M.M., Chellappan D.K., Dua K., Gupta G. Role of the Serine/Threonine Kinase 11 (STK11) or Liver Kinase B1 (LKB1) Gene in Peutz–Jeghers Syndrome. Crit Rev Eukaryot Gene Expr. 2020;30(3):245–52. doi:10.1615/CritRevEukaryotGeneExpr.2020033451. PMID:32749111.
4. Aretz S., Stienen D., Uhlhaas S., Loff S., Back W., Pagenstecher C., McLeod D.R., Graham G.E., Mangold E., Santer R., Propping P., Friedl W. High proportion of large genomic STK11 deletions in Peutz–Jeghers syndrome. Hum Mutat. 2005;26(6):513–9. doi:10.1002/humu.20253. PMID:16287113.
6. Wagner A., Aretz S., Auranen A., Bruno M.J., Cavestro G.M., Crosbie E.J., Goverde A., Jelsig A.M., Latchford A., Leerdam M.E.V., Lepisto A., Puzzono M., Winship I., Zuber V., Möslein G. The Management of Peutz–Jeghers Syndrome: European Hereditary Tumour Group (EHTG) Guideline. J Clin Med. 2021;10(3):473. doi:10.3390/jcm10030473. PMID:33513864.
7. Tomlinson I.P., Houlston R.S. Peutz–Jeghers syndrome. J Med Genet. 1997;34(12):1007–11. doi:10.1136/jmg.34.12.1007. PMID:9429144.
8. Aaltonen L.A., Jarvinen H., Gruber S.B., Billaud M., Jass J.R. Tumours of the small intestine: Peutz–Jeghers syndrome. In World Health Organization Classification of Tumours: Pathology and Genetics. Tumours of the Digestive System. IARC Press: Lyon, France, 2000.
9. Beggs A.D., Latchford A.R., Vasen H.F., Moslein G., Alonso A., Aretz S., Bertario L., Blanco I., Bülow S., Burn J., Capella G., Colas C., Friedl W., Møller P., Hes F.J., Järvinen H., Mecklin J.P., Nagengast F.M., Parc Y., Phillips R.K., Hyer W., Ponz de Leon M., Renkonen-Sinisalo L., Sampson J.R., Stormorken A., Tejpar S., Thomas H.J., Wijnen J.T., Clark S.K., Hodgson S.V. Peutz–Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–86. doi:10.1136/gut.2009.198499. PMID:20581245.
11. Traboulsi E.I., Maumenee I.H. Periocular pigmentation in the Peutz–Jeghers syndrome. Am J Ophthalmol. 1986;102(1):126–7. doi:10.1016/0002-9394(86)90229-1. PMID:3728618.
12. Jeghers H., McKusick V.A., Katz K.H. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic signifi cance. N Engl J Med. 1949;241(26):1031–6. doi:10.1056/NEJM194912292412601. PMID:15398245.
13. Цветкова Г.М., Мордовцева В.В., Вавилов А.М., Мордовцев В.Н. Патоморфология болезней кожи: Руководство для врачей. М.: Медицина, 2003. С. 320.
14. Sarkozy A., Conti E., Digilio M.C., Marino B., Morini E., Pacileo G., Wilson M., Calabrò R., Pizzuti A., Dallapiccola B. Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet. 2004;41(5):e68. doi:10.1136/jmg.2003.013466. PMID:15121796.
15. Lampe A.K., Hampton P.J., Woodford-Richens K., Tomlinson I., Lawrence C.M., Douglas F.S. Laugier–Hunziker syndrome: an important differential diagnosis for Peutz–Jeghers syndrome. J Med Genet. 2003;40(6):e77. doi:10.1136/jmg.40.6.e77. PMID:12807976.
16. Kopacova M., Tacheci I., Rejchrt S., Bures J. Peutz–Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009;15(43):5397–408. doi:10.3748/wjg.15.5397. PMID:19916169.
17. Goldstein S.A., Hoffenberg E.J. Peutz–Jeghers syndrome in childhood: need for updated recommendations? J Pediatr Gastroenterol Nutr. 2013;56(2):191–5. doi:10.1097/MPG.0b013e318271643c. PMID:23325439.
18. Hinds R., Philp C., Hyer W., Fell J.M. Complications of childhood Peutz–Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr. 2004;39(2):219–20. doi:10.1097/00005176-200408000-00027. PMID:15269641.
19. van Lier M.G., Mathus-Vliegen E.M., Wagner A., van Leerdam M.E., Kuipers E.J. High cumulative risk of intussusception in patients with Peutz–Jeghers syndrome: time to update surveillance guidelines? Am J Gastroenterol. 2011;106(5):940–5. doi:10.1038/ajg.2010.473.
21. van Lier M.G., Westerman A.M., Wagner A., Looman C.W., Wilson J.H., de Rooij F.W., Lemmens V.E., Kuipers E.J., Mathus-Vliegen E.M., van Leerdam M.E. High cancer risk and increased mortality in patients with Peutz–Jeghers syndrome. Gut. 2011;60(2):141–7. doi:10.1136/gut.2010.223750. PMID:21205875.
22. Giardiello F.M., Brensinger J.D., Tersmette A.C., Goodman S.N., Petersen G.M., Booker S.V., Cruz-Correa M., Offerhaus J.A. Very high risk of cancer in familial Peutz–Jeghers syndrome. Gastroenterology. 2000;119(6):1447–53. doi:10.1053/gast.2000.20228. PMID:11113065.
23. Boardman L.A., Pittelkow M.R., Couch F.J., Schaid D.J., McDonnell S.K., Burgart L.J., Ahlquist D.A., Carney J.A., Schwartz D.I., Thibodeau S.N., Hartmann L.C. Association of Peutz–Jeghers-like mucocutaneous pigmentation with breast and gynecologic carcinomas in women. Medicine (Baltimore). 2000;79(5):293–8. doi:10.1097/00005792-200009000-00002. PMID:11039077.
26. Amos C.I., Keitheri-Cheteri M.B., Sabripour M., Wei C., McGarrity T.J., Seldin M.F., Nations L., Lynch P.M., Fidder H.H., Friedman E., Frazier M.L. Genotype-phenotype correlations in Peutz–Jeghers syndrome. J Med Genet. 2004;41(5):327–33. doi:10.1136/jmg.2003.010900. PMID:15121768.
27. McKay V., Cairns D., Gokhale D., Mountford R., Greenhalgh L. First report of somatic mosaicism for mutations in STK11 in four patients with Peutz–Jeghers syndrome. Fam Cancer. 2016;15(1):57–61. doi:10.1007/s10689-015-9839-3.
29. Stratakis C.A., Kirschner L.S., Carney J.A. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86(9):4041–6. doi:10.1210/jcem.86.9.7903. PMID:11549623.
Синдром Пейтца-Егерса: что стало известно за 125 лет изучения? (обзор литературы)
Синдром Пейтца-Егерса (СПЕ) является крайне редким аутосомно-доминантным наследственным заболеванием, которое клинически характеризуется ростом гамартомных полипов в желудочно-кишечном тракте, слизисто-кожной пигментацией и повышенным риском возникновения злокачественных новообразований различной локализации. В большинстве случаев развитие СПЕ связано с наличием мутации в гене STK11, однако не у всех пациентов имеется данная мутация. В настоящем обзоре литературы представ- лены исторические аспекты появления данных о СПЕ, рассмотрены клинические проявления заболевания, актуальные методы диагностики, а также современные знания о генетических причинах развития СПЕ, риске возникновения злокачественных новообразований у пациентов с СПЕ, существующие рекомендации по скринингу и лечению пациентов с СПЕ. Однако наличие ряда нерешенных до настоящего времени вопросов в генетике, мониторинге и лечении свидетельствуют о необходимости дальнейших исследований.
Ключевые слова
Об авторах
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
ул. Саляма Адиля, д. 2, г. Москва, 123423, Россия
Список литературы
1. Kopacova M, Tacheci I, Rejchrt S. et al. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009;15(43):5397–5408.
2. Цуканов А.С., Шелыгин Ю.А., Фролов С.А. и соавт. Семейный аденоматоз толстой кишки. Хирург. 2017;3:14–23.
3. Шелыгин Ю.А., Кашников В.Н., Фролов С.А. и соавт. Молекулярно-генетическое исследование наследственной пред- расположенности к разным формам полипоза толстой кишки. Колопроктология. 2013;1(43):9–14.
4. Giardiello F, Trimbath J. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4(4):408–415.
5. Schreibman I, Baker M, Amos C. et al. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005 Feb;100(2):476–90.
6. McGarrity T, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci. 2006;63(18):2135–2144.
7. Hutchinson J. Pigmentation of lips and mouth. Archives of Surgery. 1896;7:290–291.
8. Peutz J. Over een zeer merkwaardige, gecombineerde familiaire polyposis van de slijmliezen van den tractus intestinalis met die van de neuskeelholte en gepaard met eigenaardige pigmentaties van huid-enslijmvliezen. Ned Maandschr Gen. 1921;10:134–146.
9. Jeghers H, McKusick V, Katz K. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241:1031–1036.
10. Bruwer A, Bargen J, Kierland R. Surface pigmentation and generalized intestinal polyposis; (Peutze-Jeghers syndrome). Proc Staff Meet Mayo Clin. 1954;29:168–171.
11. Beggs A, Latchford A, Vasen H. et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986.
14. Hinds R, Philp C, Hyer W. et al. Complications of childhood Peutz- Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr. 2004;39(2):219–20.
15. Turpin A, Cattan S, Leclerc J. et al. Prédisposition héréditaire aux cancers digestifs, mammaires, gynécologiques et gonadiques : état des lieux du syndrome de Peutz-Jeghers [Hereditary predisposition to cancers of the digestive tract, breast, gynecological and gonadal: focus on the Peutz-Jeghers]. Bull Cancer. 2014;101(9):813–822.
16. Gao H, van Lier M, Poley J. et al. Endoscopic therapy of smallbowel polyps by double-balloon enteroscopy in patients with Peutz- Jeghers syndrome. Gastrointest Endosc. 2010;71(4):768–773.
17. Vogel T, Schumacher V, Saleh A. et al. Extraintestinal polyps in Peutz-Jeghers syndrome: presentation of four cases and review of the literature. Deutsche Peutz-Jeghers-Studiengruppe. Int J Colorectal Dis. 2000;15(2):118–123.
18. Van Lier M, Mathus-Vliegen E, Wagner A. et al. High cumulative risk of intussusceptions in patients with Peutz-Jeghers syndrome: time to update surveillance guidelines? Am J Gastroenterol. 2011;106(5):940–945.
19. Tse J, Wu S, Shinagare S. et al. Peutz-Jeghers syndrome: a critical look at colonic Peutz-Jeghers polyps. Mod Pathol. 2013;26(9):1235–1240.
20. Choi H, Park Y, Youk E. et al. Clinical characteristics of Peutz- Jeghers syndrome in Korean polyposis patients. Int J Colorectal Dis. 2000;15(1):35–38.
21. Kamilaris C, Faucz F, Voutetakis A. et al. Carney Complex. Exp Clin Endocrinol Diabetes. 2019;127(2–03):156–164.
22. Sarkozy A, Conti E, Digilio M. et al. Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet. 2004;41(5):e68.
23. Aretz S, Stienen D, Uhlhaas S. et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26(6):513–519.
24. Ober W. Selected items from the history of pathology: Eugen Albrecht, MD (1872–1908): hamartoma and choristoma. Am J Pathol. 1978;91(3):606.
25. Jansen M, de Leng W, Baas A. et al. Mucosal prolapse in the pathogenesis of Peutz-Jeghers polyposis. Gut. 2006;55(1):1–5.
26. Аруин Л.И., Капуллер Л.Л., Исаков В.А. Морфологическая диагностика болезней желудка и кишечника. Москва: «Триада-Х». 1998; с. 441.
28. Hemminki A, Markie D. Tomlinson I. et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187.
29. Van Lier M, Wagner A, Mathus-Vliegen E. et al. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105(6):1258–1265.
30. Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007 Dec 13;26(57):7825–32.
32. Jishage K, Nezu J, Kawase Y. et al. Role of LKB1, the causative gene of Peutz-Jegher’s syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci U S A. 2002;99(13):8903–8908.
33. Tiainen M, Vaahtomeri K, Ylikorkala A, Mäkelä T. Growth arrest by the LKB1 tumor suppressor: induction of p21 (WAF1/CIP1). Hum Mol Genet. 2002;11(13):1497–1504.
34. Sfakianaki M, Papadaki C, Tzardi M. et al. Loss of LKB1 Protein Expression Correlates with Increased Risk of Recurrence and Death in Patients with Resected, Stage II or III Colon Cancer. Cancer Res Treat. 2019 Oct;51(4):1518–1526.
35. Schumacher V, Vogel T, Leube B. et al. STK11 genotyping and cancer risk in Peutz-Jeghers syndrome. J Med Genet. 2005;42(5):428–435.
36. Chen C, Zhang X, Wang D. et al. Genetic Screening and Analysis of LKB1 Gene in Chinese Patients with Peutz-Jeghers Syndrome. Med Sci Monit. 2016 Oct 10;22:3628–3640.
37. Vélez A, Gaitan M, Marquez J. et al. Two novel LKB1 mutations in Colombian Peutz-Jeghers syndrome patients. Clin Genet. 2009;75(3):304–306.
38. Amos C, Keitheri-Cheteri M, Sabripour M. et al. Genotypephenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004;41(5):327–333.
39. Mehenni H, Resta N, Park J. et al. Cancer risks in LKB1 germ line mutation carriers. Gut. 2006;55(7):984–990.
40. Giardiello F, Brensinger J, Tersmette A. et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453.
41. Hearle N, Schumacher V, Menko F. et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215.
42. Daniell J, Plazzer J, Perera A, Macrae F. An exploration of genotype- phenotype link between Peutz-Jeghers syndrome and STK11: a review. Fam Cancer. 2018;17:421–427.
43. Giardiello F, Welsh S, Hamilton S. et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316(24):1511–1514.
44. Utsunomiya J, Gocho H, Miyanaga T. et al. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975;136(2):71–82.
45. Van Lier M, Westerman A., Wagner A. et al. High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome. Gut. 2011;60:141–147.
46. Resta N, Pierannunzio D, Lenato G. et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013;45:606–611.
47. McGarrity T, Amos C, Baker M. Peutz-Jeghers Syndrome. 2001 Feb 23 [updated 2016 Jul 14]. In: Adam M., Ardinger H., Pagon R., Wallace S. et al. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020.
48. Chen H, Jin X, Li B. et al. Cancer risk in patients with Peutz- Jeghers syndrome: A retrospective cohort study of 336 cases. Tumour Biol. 2017;39(6):1010428317705131.
49. Taheri D, Afshar-Moghadam N, Mahzoni P. et al. Cancer problem in Peutz-Jeghers syndrome. Adv Biomed Res. 2013;2:35. 50. Bosman F. The hamartoma–adenoma–carcinoma sequence. J Pathol. 1999;188:1–2.
51. Bouraoui S, Azouz H, Kechrid H, et al. PeutzeJeghers’ syndrome with malignant development in a hamartomatous polyp: report of one case and review of the literature (In French). Gastroenterol Clin Biol. 2008;32:250–254.
52. Hizawa K, Iida M, Matsumoto T. et al. Neoplastic transformation arising in Peutz-Jeghers polyposis. Dis Colon Rectum. 1993;36:953–957.
53. Latchford A, Phillps R. Gastointestinal polyps and cancer in Peutz- Jeghers syndrome: clinical aspects. Fam Cancer. 2011;10:455–61.
54. Шелыгин Ю.А., Поспехова Н.И., Шубин В.П. и соавт. Пилотное клинико-генетическое исследование российских пациентов с син- дромом Пейтца-Егерса. Вопросы онкологии. 2016;62(1):112–116.
55. Pilleul F, Penigaud M, Milot L. et al. Possible small-bowel neoplasms: contrast-enhanced and water-enhanced multidetector CT enteroclysis. Radiology. 2006 Dec;241(3):796–801.
56. Tomas C, Soyer P, Dohan A. et al. Update on imaging of Peutz-Jeghers syndrome. World J Gastroenterol. 2014 Aug 21;20(31):10864–75.
57. Caspari R, von Falkenhausen M, Krautmacher C. et al. Comparison of capsule endoscopy and magnetic resonance imaging for the detection of polyps of the small intestine in patients with familial adenomatous polyposis or with Peutz-Jeghers’ syndrome. Endoscopy. 2004;36:1054–1059.
58. Ross A, Dye C, Prachand V. Laparoscopic-assisted double-balloon enteroscopy for small-bowel polyp surveillance and treatment in patients with Peutz-Jeghers syndrome. Gastrointestinal Endoscopy. 2006;64:984–988.
59. Postgate A, Hyer W, Phillips R. et al. Feasibility of video capsule endoscopy in the management of children with Peutzejeghers syndrome: a blinded comparison with barium enterography for the detection of small bowel polyps. J Pediatr Gastroenterol Nutr. 2009;49:417–23.
60. Su X, Ge Y. Liang B.et al. Small intestinal tumors: diagnostic accuracy of enhanced multi-detector CT virtual endoscopy. Abdom Imaging. 2012 Jun;37(3):465–74.
61. Jee Hee Son, Bo Young Chung, Min Je Junget al. Cowden Disease: Case Report and Review of the Literature. Ann Dermatol. 2019;Jun;31(3):325–330.
62. Hearle N, Schumacher V, Menko F. et al. STK11 status and intussusception risk in Peutz-Jeghers syndrome. J Med Genet. 2006;43(8):e41.
63. Spigelman A, Thomson J, Phillips R. Towards decreasing the relaparotomy rate in the Peutz-Jeghers syndrome: the role of peroperative small bowel endoscopy. Br J Surg. 1990;77(3):301–302.
64. Amaro R., Diaz G., Schneider J.et al. Peutz-Jeghers syndrome managed with a complete intraoperative endoscopy and extensive polypectomy. Gastrointest Endosc. 2000;52(4):552–554.
65. Edwards D, Khosraviani K, Stafferton R, Phillips R. Long-term results of polyp clearance by intraoperative enteroscopy in the Peutz-Jeghers syndrome. Dis Colon Rectum. 2003;46(1):48–50.
66. Sakamoto H, Yamamoto H, Hayashi Y. et al. Nonsurgical management of small-bowel polyps in Peutz-Jeghers syndrome with extensive polypectomy by using double-balloon endoscopy. Gastrointest Endosc. 2011;74(2):328–333.
67. Robinson J, Lai C, Martin A. et al. Oral rapamycin reduces tumour burden and vascularization in LKB1+/– mice. J Pathol. 2009;219(1):35–40.
68. Kuwada S, Burt R. A rationale for mTOR inhibitors as chemoprevention agents in Peutz-Jeghers syndrome. Fam Cancer. 2011;10(3):469–472.
69. Ishida H, Tajima Y, Gonda T. et al. Update on our investigation of malignant tumors associated with Peutz-Jeghers syndrome in Japan. Surg Today. 2016 Nov;46(11):1231–42.
Синдром Пейтца-Егерса
Появление пигментных пятен кофейного цвета в области губ и слизистой оболочки рта может свидетельствовать о наличии редкого онкологического синдрома.
На консультацию к врачу-генетику Центра персонализированной медицины МКНЦ обратилась пациентка М. в возрасте 35 лет с диагнозом множественный полипоз толстой кишки, рак толстой кишки. Из активных жалоб отмечалась общая слабость, недомогание, периодические боли в животе и нарушение стула на протяжении длительного времени.
За несколько месяцев до этого пациентка обнаружила кровь в стуле, после чего обратилась за помощью в поликлинику по месту жительства, где при колоноскопии выявлено более 50 полипов толстой кишки и заподозрено злокачественное новообразование. Пациентка сообщила, что ее мать умерла от рака толстой кишки в возрасте 40 лет. При осмотре обращала на себя внимание светлая фарфоровая кожа с множеством веснушчатых пигментных пятен, равномерно расположенных по всему телу, рыжий цвет волос и светлый оттенок глаз. Однако скопление пигментаций в области губ усиливалось, они переходили на слизистую оболочку полости рта и щек, где размеры пятен достигали 5 мм в диаметре. Пациентка пришла на прием с родной сестрой, у которой подобных особенностей не наблюдалось.
Учитывая данные осмотра, отягощенный семейный анамнез раком толстой кишки и выявление полипов при колоноскопии у пациентки заподозрен синдром Пейтца-Егерса, в связи с чем наши специалисты рекомендовали проведение молекулярно-генетической диагностики для исключения наследственной природы заболевания и разработали индивидуальный план наблюдения.
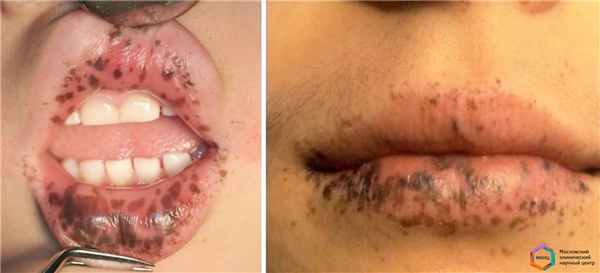
Синдром Пейтца-Егерса – это редкий наследственный онкологический синдром, который развивается в результате нарушений в строении гена STK11. Классически этот синдром проявляется скоплением множества пигментных пятен кофейного цвета размерами до 5-7 мм. Преимущественно они располагаются на губах, слизистой оболочке рта, в некоторых случаях на кончиках пальце или стопах и наблюдаются с детства. Второй особенностью данного синдрома является наличие множества полипов (доброкачественных новообразований) желудочно-кишечного тракта, которые способны к злокачественной трансформации. Наиболее часто полипы выявляются в тонкой кишке, но также могут встречаться в желудке и толстой кишке. На протяжении жизни они могут изъязвляться, кровоточить, вызывать кишечную инвагинацию и непроходимость, что приводит к установлению диагноза в возрасте до 20 лет у 50% больных.
Известно, что у пациентов с синдромом Пейтца-Егерса риски возникновения злокачественных новообразований в различных органах в несколько раз выше, чем без мутации в гене STK11. Пациенты с данным синдромом нуждаются в составлении индивидуального плана периодического наблюдения, в который обязательно входит колоноскопия, маммография, эзофагогастродуоденоскопия, эндоскопическое исследование тонкой кишки, гинекологическое обследование у женщин. В случае обнаружения подозрительных полипов специалисты рекомендуют их эндоскопическое иссечение - полипэктомия.
Учитывая, что риски наследования данной особенности составляют 50%, при обнаружении мутации у пациента рекомендуется консультация врача-генетика и проведение молекулярно-генетической диагностики у его родственников для своевременного оказания специализированной медицинской помощи.
Синдром Пейтца–Егерса: диагностические и лечебные возможности современной внутрипросветной эндоскопии на примере собственного клинического наблюдения
Синдром Пейтца–Егерса — достаточно редкое заболевание с аутосомно-доминантным типом наследования, характеризующееся множественными гамартомами в желудочно-кишечном тракте. Лечение таких больных традиционно хирургическое, но в настоящее время благодаря внедрению в клиническую практику новейших методик внутрипросветной эндоскопии зачастую удается избежать открытых полостных операций и провести полноценную диагностику и удаление гамартомных полипов минимально инвазивным путем. Целью публикации данной статьи стала необходимость на примере собственного клинического наблюдения продемонстрировать диагностические и оперативные возможности современной внутрипросветной эндоскопии в лечении пациентов с множественным наследственным полипозом желудочно-кишечного тракта. Методика глубокой однобаллонной энтероскопии в сочетании со стандартными рутинными эндоскопическими манипуляциями позволяет выполнить тотальный осмотр всего желудочно-кишечного тракта и в случае необходимости провести удаление практически любых эпителиальных неоплазий, что, безусловно, положительно сказывается на качестве жизни пациентов.
Ключевые слова
Об авторах
Научный центр здоровья детей; Первый Московский государственный медицинский университет им. И.М. Сеченова
Россия
доктор медицинских наук, профессор кафедры детской хирургии и урологии-андрологии, врач высшей категории, заведующий отделением эндоскопических и морфологических исследований НЦЗД,
119991, Москва, Ломоносовский проспект, д. 2, стр. 1
Список литературы
1. Кайбышева В.О., Ивашкин В.Т., Баранская Е.К., и др. Синдром Пейтца–Егерса: обзор литературы и описание собственного клинического наблюдения // Российский журнал гастроэнтерологии, гепатологии, колопроктологии. — 2011. — Т. 21. — № 2 — С. 54–61. [Kaibysheva VO, Ivashkin VT, Baranskaya EK, et al. Sindrom Peittsa–Egersa: obzor literatury i opisanie sobstvennogo klinicheskogo nablyudeniya. Russian journal of gastroenterology, hepatology, coloproctology. 2011;21(2):54–61. (In Russ).]
2. Цуканов А.С., Шубин В.П., Поспехова Н.И., и др. Наследствен ные раки желудочно-кишечного тракта // Практическая онкология. — 2014. — Т.15. — №3 — С. 126–133. [Tsukanov AS, Shubin VP, Pospekhova NI, et al. Nasledstvennye raki zheludochnokishechnogo trakta. Prakticheskaya onkologiya. 2014;15(3): 126–133. (In Russ).]
4. Iacobuzio-Donahue CA, Montgomery EA. Gastrointestinal and liver pathology. A volume in the series: Foundations in diagnostic pathology. 2nd ed. Philadelphia: Saunders Elsevier; 2011. 736 p.
5. Basak F, Kinaci E, Aksoy S, et al. Multiple intestinal intussusceptions in Peutz–Jeghers’ syndrome: A case report. Acta Chir Belg. 2010;110(1):93–94. doi: 10.1080/00015458.2010.11680575.
6. Jansen M, Leng W, Baas AF, et al. Mucosal prolapse in the pathogenesis of Peutz–Jeghers polyposis. Gut. 2006;55(1):1–5. doi: 10.1136/gut.2005.069062.
8. Казубская Т.П., Белев Н.Ф., Козлова В.М., и др. Наследственные синдромы, ассоциированные с полипами и развитием злокачественных опухолей у детей // Онкопедиатрия. — 2015. — Т. 2. — № 4 — С. 384–395. [Kazubskaya TP, Belev NF, Kozlova VM, et al. The hereditary syndromes associated with polyps and development of malignant tumours in children. Oncopediatria. 2015;2(4):384–395. doi: 10.15690/onco.v2.i4.1465. (In Russ).] doi: 10.15690/onco.v2.i4.1465
9. Beggs AD, Latchford AR, Vasen HF, еt al. Peutz–Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986. doi: 10.1136/gut.2009.198499.
Синдром Пейтца–Егерса
Синдром Пейтца–Егерса (англ. Peutz–Jeghers syndrome) или гамартомный полипоз желудочно-кишечного тракта (ЖКТ) — орфанное (Запруднов А.М.) заболевание, характеризующееся множественными доброкачественными полипами (гамартомами) в желудке, тонкой и толстой кишке и, одновременно, пигментными пятнами тёмно-коричневого цвета, круглой или овальной формы, диаметром от 1 до 5 мм, располагающимися обычно на коже лица, вокруг рта и глаз, на носу, реже на конечностях, на слизистой оболочке полости рта, иногда гениталий и прямой кишки.
Синдром Пейтца–Егерса — заболевание, наследуемое по аутосомно-доминантному типу. Распространенность его, по данным разных авторов, составляет от 1 к 50 000 до 1 к 200 000 новорожденных. Частота заболевания одинакова для лиц мужского и женского полов.
Меланиновая пигментация губ и слизистой оболочки щёк при синдроме Пейтца-Егерса (Кайбышева В.О. и др.)
По Международной классификации болезней МКБ-10 синдром Пейтца–Егерса относится к «Классу XVII. Врожденные аномалии [пороки развития], деформации и хромосомные нарушения (Q00-Q99)», блоку «Q80-Q89 Другие врожденные аномалии», рубрике «Q85.8 Другие факоматозы, не классифицированные в других рубриках».
Полипы при синдроме Пейтца-Егерса обнаруживаются на протяжении всего желудочно-кишечного тракта: от желудка до прямой кишки. Обычно располагаются неравномерно: мелкие чаще локализуются в желудке, двенадцатиперстной и толстой кишке, а крупные — в тощей. Макроскопически полипы могут быть плоскими или высокими, различной величины (от нескольких миллиметров до 5 и более сантиметров), с гладкой либо дольчатой поверхностью, на широком основании и на ножке. Полипы располагаются по одиночке или кластерами, порой выстилая всю поверхность слизистой, напоминая ковровое покрытие. У пациентов с синдромом Пейтца–Егерса наряду с гамартомными полипами в толстой кишке и желудке часто обнаруживаются аденоматозные полипы (Кайбышева В.О. и др.).
Согласно теории эмбриональной дистопии полипы при синдроме Пейтца-Егерса рассматриваются как результат неправильного эмбрионального развития слизистой оболочки желудка. Они сохраняются в слизистой оболочке желудка с эмбрионального периода и обладают высокой потенциальной энергией роста (Эрдес С.И., Сергеева Т.Н.).
Синдром Пейтца–Егерса является так называемым «вероятно предраковым» заболеванием, с риском развития рака от 5 до 10 % (Тертычный А.С.).
Молекулярно-генетическая основа синдрома Пейтца–Егерса
Причина синдрома — герминальная мутация в генах LKB1 или STK11. Мутация этого гена приводит к сокращению длины белка и утрате киназной активности. Основные типы мутаций: небольшие делеции/инсерции, нонсенс, миссенс или большие делеции, приводящие к преждевременной терминации синтеза белка.
Мутации в гене STK11 определяются у 70–80% пациентов. Причина различий в диагностике мутаций бывает обусловлена методами, используемыми для их выявления. Кроме того, для пациентов с фенотипом синдрома, но без выявленных мутаций в гене STK11, обсуждается гетерогенность и возможность существования второго гена, ответственного за синдром в локусе 19q13.4 (Казубская Т.П. и др.).
Читайте также: