Дефицит альфа-1 антитрипсина
Добавил пользователь Владимир З. Обновлено: 27.01.2026
Альфа-1-антитрипсин (ААТ) – это белок, производимый печенью и высвобождаемый в кровяное русло. Он принимает участие в инактивации нескольких ферментов, но главная его функция – защита легких от эластазы.
Синонимы русские
Синонимы английские
Alpha1-antitrypsin, A1AT, AAT.
Метод исследования
Единицы измерения
Г/л (грамм на литр).
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не принимать пищу в течение 12 часов до исследования.
- Исключить физическое и эмоциональное перенапряжение и не курить в течение 30 минут перед исследованием.
Общая информация об исследовании
Альфа-1-антитрипсин – белок, который вырабатывается печенью. Он помогает организму в инактивации ферментов, при этом основная его функция состоит в защите легких от эластазы – она производится нейтрофилами в ответ на повреждения и воспаления. Эластаза расщепляет белки, которые затем перерабатываются организмом и удаляются. Если ее активность не контролируется альфа-1-антитрипсином, она начинает разрушать ткани легких.
Синтез альфа-1-антитрипсина регулируется двумя копиями гена протеазного ингибитора серпина-1. Это так называемый кодоминантный ген, то есть каждая копия гена серпина-1 отвечает за образование половины гена альфа-1-антитрипсина. При изменениях или мутациях одной или обеих копий гена образуется меньшее количество альфа-1-антитрипсина либо его дисфункциональная разновидность. Если в результате этого продукция альфа-1-антитрипсина падает более чем на 30 % ниже нормы, то наступает расстройство, называемое дефицитом альфа-1-антитрипсина. При этом повышается риск возникновения эмфиземы, а также болезней легких в начале полового созревания. Курение и регулярный контакт с дымом и пылью ускоряют развитие болезни и усложняют ее течение из-за повреждения легких.
Дисфункциональный альфа-1-антитрипсин откладывается в клетках печени, производящих его. По мере накопления дефектный альфа-1-антитрипсин образует аномальные белковые цепи, которые начинают разрушать клетки и повреждать печень. Около 10 % больных, подверженных альфа-1-антитрипсиновой недостаточности, страдали от желтухи, будучи еще младенцами. Большинство из них поправляется, однако в тяжелых случаях больным детям для выживания требуется пересадка печени. В настоящее время альфа-1-антитрипсиновая недостаточность является наиболее распространенной болезнью печени в педиатрии.
Количество производимого альфа-1-антитрипсина и его активность зависят от типа унаследованной мутации. Несмотря на то что ген серпин-1 есть более чем в 75 аллелях, лишь несколько из них наиболее распространены. Чаще других встречаются дефектные формы гена S и Z. Существуют различные варианты их наследования.
- Одна копия М и одна копия S или Z (MS или MZ). В этом случае количество альфа-1-антитрипсина хотя и пониженное, но достаточное для защиты организма. Пациенты с таким сочетанием генов являются носителями болезни и могут передать ее по наследству своим детям.
- Две копии S (SS) обычно не приводят к клинически выраженному функциональному дефициту антитрипсина либо обуславливают лишь умеренное уменьшение его синтеза (образуют около 60 % необходимого альфа-1-антитрипсина).
- Одна копия S и одна Z (SZ) повышают риск возникновения эмфиземы (образуется около 40 % альфа-1-антитрипсина от нормального количества).
- Две копии Z (ZZ) являются причиной наиболее тяжелой формой болезни (образуется лишь около 10 % необходимого альфа-1-антитрипсина). Если такой вариант наследования сочетается с наследованием двух редких копий гена серпина-1, то возникает так называемая нулевая разновидность гена, при которой альфа-1-антитрипсин не образуется совсем.
Для определения уровня альфа-1-антитрипсина, а также для выяснения, какие аллели гена серпина-1 имеются у пациента, применяют следующие методики.
- Анализ на альфа-1-антитрипсин выявляет уровень этого белка в организме.
- Определение фенотипа генов, ответственных за синтез альфа-1-антитрипсина, позволяет выявить образующиеся формы белка альфа-1-антитрипсина и сравнить их с известными изоформами.
- Анализ ДНК-последовательности генов, связанных с образованием альфа-1-антитрипсина, помогает узнать вид мутации гена протеазного ингибитора (серпина-1). Обычно определяются только наиболее распространенные мутации (M, S, Z). Генетический анализ может применяться как для обследования больных пациентов, так и для проверки членов их семей.
Для чего используется исследование?
Для диагностики причин эмфиземы, особенно если пациент не подвержен таким факторам риска, как курение или регулярный контакт с раздражающими веществами типа пыли и дыма.
Для выявления причин продолжительной желтухи и других нарушений функции печени (главным образом у детей и подростков).
Когда назначается исследование?
- Если желтуха у новорождённого или малолетнего ребенка длится дольше 1-2 недель, при этом у него есть признаки поражения печени (увеличение селезенки, брюшная водянка, зуд).
- Когда пациент моложе 40 лет жалуется на хрипы, хронический кашель или бронхит, тяжелую одышку после физических нагрузок, а также на другие симптомы эмфиземы. Это особенно важно, когда человек не курит, не контактирует с раздражителями легких и при этом у него диагностировано повреждение нижней части легких.
- Если у пациента имеется близкий родственник, страдающий от альфа-1-антитрипсиновой недостаточности.
Что означают результаты?
Референсные значения: 0,9 - 2 г/л.
Чем ниже уровень альфа-1-антитрипсина, тем выше риск возникновения эмфиземы.
Опасность последствий образования дефектных форм альфа-1-антитрипсина зависит от общего количества таких форм и от разновидности аномальных генов, кодирующих альфа-1-антитрипсин. Это может быть как эмфизема вследствие недостаточной защиты легких, так и заболевание печени из-за накопления в ней дисфункциональных форм этого белка.
Образование меньшего количества альфа-1-антитрипсина связано с наличием одной или двух аномальных копий гена серпина-1. При этом степень дефицита альфа-1-антитрипсина, а также уровень повреждения легких и/или печени могут сильно варьироваться. У двух пациентов, имеющих одну и ту же дефектную копию гена, болезнь иногда протекает совершенно по-разному.
Изменение уровня альфа-1-антитрипсина происходит при острых и хронических воспалительных заболеваниях, инфекциях, а также при некоторых опухолях.
Концентрация альфа-1-антитрипсина иногда понижается у новорождённых при респираторных заболеваниях, а также при уменьшении количества общего белка из-за, например, поражения почек (нефротического синдрома), недостаточного питания и некоторых видов рака.
Что может влиять на результат?
Показатель анализа может повышаться из-за приема оральных контрацептивов, беременности, стресса и нарушений работы щитовидной железы.
Важные замечания
Дополнительно рекомендуется проведение генетического теста для выявления мутаций в гене серпина-1:
Дефицит Альфа-1-антитрипсина
Дефицит альфа 1-антитрипсина (A1AD), синдром Laurell-Eriksson - это генетическое заболевание, сопровождающееся нарушением образования альфа-1-антитрипсина (A1AT) (уменьшением его количества или изменением структуры). Альфа1-антитрипсин - ингибитор нейтрофильной эластазы (антипротеаза), главная функция которого - защитить легкие от деструкции (разрушения) ткани протеазами. Большая часть альфа1-антитрипсина синтезируется клетками печени и моноцитами и распространяется через кровоток в легкие; некоторая часть производится альвеолярными макрофагами и эпителиоцитами легких. Печеночное накопление аберрантных (измененных) молекул альфа1-антитрипсина вызывает холестатическую желтуху новорожденных у 10-20 % пациентов. В легком дефицит альфа1-антитрипсина увеличивает активность нейтрофильной эластазы, это способствует деструкции легочной ткани и развитию эмфиземы (особенно у курильщиков, так как дым сигарет также увеличивает протеазную активность). Дефицит альфа1-антитрипсина, как считается, является причиной 1-2 % всех случаев ХОБЛ – хронических обструктивных заболеваний. Другие нарушения, возможно связанные с вариантами альфа1-антитрипсина, включают панникулит; опасное для жизни кровотечение (за счет мутации альфа1-антитрипсина, которая перенаправляет ингибирующий эффект с нейтрофильной эластазы на фактор коагуляции); аневризмы; неспецифический язвенный колит и гломерулонефриты.
A1AD был описан в 1963 году Карлом-Бертиль Лорелом (Laurell Carl-Bertil), работавшим в Университете города Лунд (Швеция). К.Б.Лорел вместе с Эриксоном С. (Eriksson Sture) заметил α1 группы в структуре белка при электрофорезе в пяти из 1500 образцов крови. Как позже было установлено, у трех из пяти пациентов, которым принадлежали эти образцы, в анамнезе имела место эмфизема, возникшая в молодом возрасте. Связь дефицита А1АТ с заболеваниями печени была установлена Шарпом (Sharp и др.) шесть лет спустя.
Поражение печени у младенцев проявляется холестатической желтухой и гепатомегалией в течение первой недели жизни; желтуха обычно исчезает по истечении двух- или четырехмесячного возраста. Приблизительно в 20 % случаев поражения печени у новорожденных приводят к развитию цирроза в детстве. У 10 % пациентов, у которых в детстве не было заболевания печени, развивается цирроз печени во взрослом возрасте. Вовлечение печени увеличивает риск возникновения рака печени.
Панникулит - воспалительное заболевание тканей подкожно жировой клетчатки - проявляется в виде индурированных (уплотненных), обесцвеченных пятен или узелков, обычно на нижней части брюшной стенки, ягодицах и бедрах.
Дефицит альфа1-антитрипсина обычно вызывает раннюю эмфизему; симптомы заболевания - такие же, как при ХОБЛ. Вовлечение в процесс легких встречается раньше у курильщиков, чем у некурящих, но в обоих случаях редко развивается до возраста 25 лет. Тяжесть поражения легких очень варьирует; легочная функция хорошо сохраняется у некоторых курильщиков и может быть серьезно ухудшена у некурящих. Обструкция дыхательных путей встречается более часто у мужчин и людей с бронхиальной астмой, повторными инфекциями дыхательных путей, профессиональными воздействиями пыли и семейным анамнезом легочных заболеваний. Самая частая причина смерти при дефиците альфа1-антитрипсина - эмфизема, сопровождаемая циррозом, раком печени.
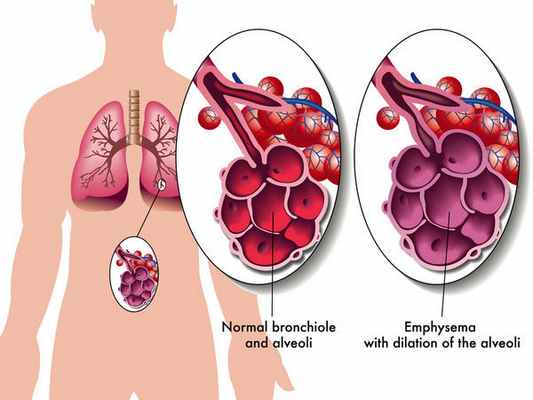
Дефицит альфа1-антитрипсина подозревается:
- у курильщиков, с развившейся эмфиземой легких до 45 лет;
- у некурящих без профессиональных вредностей, с развитием эмфиземы в любом возрасте;
- у больных с эмфиземой преимущественно в нижних долях (по данным Rg грудной клетки);
- у больных с семейным анамнезом эмфиземы или необъясненного цирроза;
- у больных с панникулитом;
- у новорожденных с желтухой или повышением ферментов печени;
- у любого пациента с необъясненным заболеванием печени.
Диагноз подтверждается исследованием сывороточного уровня альфа1-антитрипсина (< 80 мг/дл или < 11 мкмоль/л).
Дефицит альфа-1-антитрипсина, SERPINA1, ч.м.
Дефицит альфа-1-антитрипсина является наследственным заболеванием, связанным с дефектом гена SERPINA1, который приводит к патологии легких и печени.
Сывороточный белок альфа-1-антитрипсин (А1АТ) выполняет функцию ингибитора сериновых протеаз и относится к группе острофазных белков. Основой его функцией является контроль протеолиза в рыхлой соединительной ткани нижних дыхательных путей. При дефиците А1АТ структура альвеол легких разрушается, формируется эмфизема и бронхоэктазы. Основным механизмом повреждения печени является накопление дефектного A1AT и блок его секреции из ЭПС гепатоцитов. Это ведет к накоплению белковых агрегатов, в результате которого клетка погибает апоптозом.
Заболевание поражает 1 из 1 500 – 3 500 человек. Нередко дефицит А1АТ протекает под «масками» хронической обструктивной болезни легких (ХОБЛ) и резистентной бронхиальной астмы, так и оставаясь не диагностированным.
Первые признаки заболевания легких у людей с дефицитом А1АТ обычно появляются в возрасте от 20 до 50 лет. Самыми ранними симптомами являются снижение толерантности к физической нагрузке и одышка. Другие проявления могут включать необъяснимую потерю веса, рецидивирующие респираторные инфекции, учащенное сердцебиение и формирование эмфиземы. К основным чертам эмфиземы легких относят затрудненное дыхание, кашель и бочкообразную форму грудной клетки. У некоторых пациентов изменения происходят в печени - формируется хронический гепатит и цирроз. К признакам цирроза относят увеличение объема живота (асцит), отеки нижних конечностей и прогрессирующую желтуху.
Наиболее информативным методом исследования для постановки диагноза дефицита А1АТ является молекулярно-генетическое исследование гена SERPINA1. Наиболее распространены в популяции 2 мутации гена - G264V и G342L, ассоциированные с PiS- и PiZ-фенотипами соответственно.
Большинство пациентов с поражением печени или легких являются гомозиготными по PiZ- или PiS- фенотипу или являются гетерозиготными по двум аллелям. У таких больных уровень А1АТ в сыворотке снижается до 40–60%. Дефицит А1АТ наследуется по аутосомно-доминантному типу. При обнаружении этих мутаций пациенту должно быть рекомендовано генетическое консультирование ближайших кровных родственников на предмет выявления носителей.
Cпециальной подготовки не требуется. Взятие крови желательно проводить не ранее 4 часов после приема пищи.
- Диагностика дефицита альфа-1-антитрипсина;
- Дифференциальная диагностика эмфиземы лёгких и бронхоэктазов;
- Дифференциальная диагностика хронической обструктивной болезни легких (ХОБЛ) у лиц младше 40 лет и устойчивой к терапии бронхиальной астмы;
- Некротизирующий панникулит и АNCА-ассоциированный васкулит;
- Дифференциальная диагностика фиброза и цирроза печени;
- Наличие в семейном анамнезе эмфиземы легких, бронхоэктазов, болезни печени, панникулита, либо подтвержденный дефицит А1АТ у кровных родственников.
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Обнаружение гомозиготного носительства мутаций в гене SERPINA1 (генотипы SS и ZZ) или совместное наличие двух гетерозиготных мутаций (генотип SZ) свидетельствует о дефиците альфа-1-антитрипсина. Гетерозиготное носительство одной из мутаций (генотипы MS и MZ) может предрасполагать к развитию умеренной клинической картины дефицита альфа-1-антитрипсина или обуславливать скрытое носительство.
Отсутствие двух наиболее распространенных мутаций в гене SERPINA1 практически исключает диагноз дефицит альфа-1-антитрипсина. Пациентам с генотипом MM и выраженной клинической картиной заболевания может быть рекомендовано исследование гена SERPINA1 на предмет наличия более редких мутаций.
Дефицит альфа-1-антитрипсина у детей: описание серии случаев
Дефицит альфа-1-антитрипсина (А1АТ) — причина орфанного заболевания, случаи которого достаточно хорошо описаны у взрослых пациентов, однако у детей они освещены лишь в единичных публикациях, при этом зачастую ограничены описанием поражений печени. В настоящей статье представлены результаты наблюдения 5 детей с дефицитом альфа-1-антитрипсина, в числе которых 3 мальчика (гомозиготы по Z-аллели) и 2 девочки (носители PiMZ-фенотипа). Показано, что поражения легочной ткани у пациентов с дефицитом А1АТ дебютировали в возрасте 2 лет с признаками рецидивирующей бронхиальной обструкции и в 7 лет в виде эмфиземы легких. Повышение осведомленности практикующих врачей различных специальностей позволит улучшить диагностику указанной формы патологии и коморбидных с ней состояний.
Ключевые слова
Об авторах
Северо-Западный государственный медицинский университет им. И.И. Мечникова Детская городская больница № 19 им. К.А. Раухфуса
Россия
ассистент кафедры педиатрии и детской кардиологии СЗГМУ им. И.И. Мечникова Адрес: 191015, Санкт-Петербург, ул. Кирочная, д. 41
Санкт-Петербургский государственный педиатрический медицинский университет
Россия
Санкт-Петербург, Российская Федерация
Северо-Западный государственный медицинский университет им. И.И. Мечникова Детская городская больница № 19 им. К.А. Раухфуса
Россия
Санкт-Петербург, Российская Федерация
Северо-Западный государственный медицинский университет им. И.И. Мечникова Детская городская больница № 19 им. К.А. Раухфуса
Россия
Санкт-Петербург, Российская Федерация
Северо-Западный государственный медицинский университет им. И.И. Мечникова
Россия
Санкт-Петербург, Российская Федерация
Список литературы
1. Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397–415.
2. Основы клинической гепатологии. Заболевания печени и билиарной системы. Учебное пособие для системы последипломного образования врачей / Под ред. В.Г. Радченко, А.В. Шаброва, Е.Н. Зиновьева. — СПб.: Диалект; М.: БИНОМ; 2005. — 862 с. [Osnovy klinicheskoi gepatologii. Zabolevaniya pecheni i biliarnoi sistemy. Uchebnoe posobie dlya sistemy poslediplomnogo obrazovaniya vrachei. Ed by Radchenko V.G., Shabrov A.V., Zinov’ev E.N. St. Petersburg: Dialekt; Moscow: BINOM; 2005. 862 p. (In Russ).]
3. Жигальцова-Кучинская А., Сивицкая Л.Н., Даниленко Н.Г. и др. Дефицит альфа-1- антитрипсина: генетические основы, эпидемиология, значение в развитии бронхо- легочной патологии // Вестник Витебского государственного медицинского университета. — 2015. — Т. 14. — № 6 — С. 39–52. [Zhigaltsova-Kuchinskaya OA, Sivitskaya LN, Danilenko NG, et al. Alpha-1-antitrypsin deficiency: genetic fundamentals, epidemiology, role in the development of bronchopulmonary pathology. Vestnik Vitebskogo gosudarstvennogo meditsinskogo universiteta. 2015;14(6):39–52. (In Russ).]
4. Колесникова Е.В. Альфа-1-антитрипсиновая недостаточность: Современный взгляд на проблему // Сучасна гастроентерологія. — 2008. — № 2 — С. 93–98. [Kolesnikova EV. Al’fa-1-antitripsinovaya nedostatochnost’: Sovremennyi vzglyad na problemu. Contemporary gastroenterology. 2008;(2):93–98. (In Russ).]
6. Churg A, Wang X, Wang RD, et al. 1-Antitrypsin suppresses TNF- and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144– 151. doi: 10.1165/rcmb.2006-0345OC.
7. Sharp HL. Alpha-1-antitrypsin deficiency. Hosp Pract. 1971; 6(5):83–96. doi: 10.1080/21548331.1971.11706032.
11. Laurell CB, Eriksson S. The electrophoretic pattern alpha-1-globulin pattern of serum in alpha-l-antitrypsin deficiency. Scan J Clin Lab Invest. 1963;15(2):132–140. doi: 10.3109/00365516309051324.
12. Pariente EA, Degott C, Martin JP, et al. Hepatocytic PAS-positive diastase-resistance inclusions in the absence of alpha-1-antitrypsin deficiency — high prevalence in alcoholic cirrhosis. Am J Clin Pathol. 1981;76(3):299-302. doi: 10.1093/ajcp/76.3.299.
13. Протеолиз в норме и при патологии / Под ред. К.Н. Веремеенко, О.П. Голобородько, А.И. Кизим. — Киев: Здоров’я; 1988. — 199 с. [Proteoliz v norme i pri patologii. Ed by Veremeenko K.N., Goloborod’ko O.P., Kizim A.I. Kiev: Zdorov’ya; 1988. 199 p. (In Russ).]
15. Веремеенко К.Н. Ферменты протеолиза и их ингибиторы в медицинской практике. — Киев; 1971. — 216 с. [Veremeenko KN. Fermenty proteoliza i ikh ingibitory v meditsinskoi praktike. Kiev; 1971. 216 p. (In Russ).]
16. Hubbard RC, Crystal RG. Strategies for aerosol therapy of alpha 1-antitrypsin deficiency by the aerosol route. Lung. 1990;168 Suppl:565–578. doi: 10.1007/bf02718179.
17. Crowther DC, Belorgey D, Miranda E, et al. Practical genetics: alpha-1-antitrypsin deficiency and the serpinopathies. Eur J Hum Genet. 2004;12(3):167–172. doi: 10.1038/sj.ejhg.5201127.
18. Шапошникова Н.А., Шулятьев И.С., Варванина Г.Г., Дроздов В.Н. Клиническое значение наследственного и приобретенного дефицита альфа-1-антитрипсина у больных циррозом печени и болезнью Вильсона-Коновалова // Лабораторная служба. — 2010. — № 10 — С. 12–16. [Shaposhnikova NA, Shulyat’ev IS, Varvanina GG, Drozdov VN. Klinicheskoe znachenie nasledstvennogo i priobretennogo defitsita al’fa-1-antitripsina u bol’nykh tsirrozom pecheni i bolezn’yu Vil’sona-Konovalova. Laboratornaya sluzhba. 2010; (10):12–16. (In Russ).]
19. Бродская О.Н. Наследственная недостаточность 1-антитрипсина // Практическая пульмонология. — 2008. — № 4 — С. 58–59. [Brodskaya ON. Nasledstvennaya nedostatochnost’ 1-antitripsina. Prakticheskaya pul’monologiya. 2008;(4):58–59. (In Russ).]
20. Аверьянов А.В., Поливанова А.Э. Дефицит 1-антитрипсина и хроническая обструктивная болезнь легких // Пульмонология. — 2007. — № 3 — С. 103–109. [Averyanov AV, Polivanova AE. Alfa-1-antitripsin deficiency and chronic obstrictivc pulmonary disease. Pul’monologiya. 2007;(3):103–109. (In Russ).]
21. Castaldi PJ, DeMeo DL, Kent DM, et al. Development of predictive models for airflow obstruction in alpha-1-antitrypsin deficiency. Am J Epidemiol. 2009;170(8):1005–1013. doi: 10.1093/aje/kwp216.
22. Davis ID, Burke B, Freese D, et al. The pathologic spectrum of the nephropathy associated with 1-antitrypsin deficiency. Hum Pathol. 1992;23(1):57–62. doi: 10.1016/0046- 8177(92)90012-r.
23. Churg A, Wang X, Wang RD, et al. Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Respir Cell Mol Biol. 2007;37(2):144–151. doi: 10.1165/rcmb.2006-0345OC.
24. Видаль Р., Бланко И., Касас Ф. и др. Рекомендации по диагностике и ведению больных с дефицитом 1-антитрипсина Испанского общества пульмонологии и торакальной хирургии (SEPAR) // Пульмонология. — 2008. — № 1 — С. 14–28. [Vidal R, Blanco I, Casas F, et al. The National Alpha-1 Antitrypsin Registry Committee Guidelines for the diagnosis and management of alpha-1-antitrypsin deficiency of SEPAR. Pul’monologiya. 2008;(1):14–28. (In Russ).]
25. Назаров П.Г. Реактанты острой фазы воспаления.— СПб.: Наука; 2001. — 423 с. [Nazarov PG. Reaktanty ostroi fazy vospaleniya. St. Petersburg: Nauka; 2001. 423 p. (In Russ).]
26. Tanash HA, Nilsson PM, Nilsson JA, et al. Survival in severe alpha-1-antitrypsin deficiency (PiZZ). Respir Res. 2010;11:44. doi: 10.1186/1465-9921-11-44.
27. Bornhorst J, Calderon F, Procter M, et al. Genotypes and serum concentrations of human alpha-1-antitrypsin “P” protein variants in a clinical population. J Clin Pathol. 2007;60(10):1124–1128. doi: 10.1136/jcp.2006.042762.
29. Elzouki AN, Segelmark M, Wieslander J, Eriksson S. Strong link between the alpha 1- antitrypsin PiZ allele and Wegener’s granulomatosis. J Intern Med. 1994;236(5):543–548. doi: 10.1111/j.1365-2796.1994.tb00842.x.
30. Askari FK. Molecular mechanism of hepatocellular injury in alpha 1 antitrypsin deficiency. Hepatology. 1995;21(6):1745–1747. doi: 10.1002/hep.1840210638.
31. Strange C, Dickson R, Carter C, et al. Genetic testing for alpha1-antitrypsin deficiency. Genet Med. 2004;6(4):204–210. doi: 10.109701.GIM.0000132669.09819.79.
32. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. doi: 10.1164/ajrccm.153.2.8564092.
33. Fregonese L, Stolk J. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis. 2008;3:16. doi: 10.1186/1750-1172-3-16.
34. McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1- Antitrypsin Deficiency Registry Study Group. Chest. 1997;111(2):394–403. doi: 10.1378/chest.111.2.394.
35. Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: Clinical manifestations and natural history. Thorax. 2004;59(5): 441–445. doi: 10.1136/thx.2003.006510.
36. Katz RM, Lieberman J, Siegel SC. Alpha-1 antitrypsin levels and prevalence of Pi variant phenotypes in asthmatic children. J Allergy Clin Immunol. 1976;57(1):41–45. doi: 10.1016/0091-6749(76)90077-4.
37. Bruttmann G. [Reagin asthma and familial alpha-1antitrypsin deficiency. (In French).] Nouv Presse Med. 1974;3(10):589–591.
38. Bruttmann G. [Asthma associated with a familial deficiency in alpha-1-antitrypsin. (In French).] Revue francaise d’allergologie. 1973;13(4):411–418 doi: 10.1016/S0035- 2845(73)80062-9.
39. Browne RJ, Mannino DM, Khoury MJ. Alpha 1-antitrypsin deficiency deaths in the United States from 1979–1991. An analysis using multiple-cause mortality data. Chest. 1996;110(1):78–83. doi: 10.1378/chest.110.1.78.
40. Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294(24): 1316–1321. doi: 10.1056/NEJM197606102942404.
Недостаточность альфа-1 антитрипсина
При данном наследственном заболевании печени в крови либо отсутствует либо значительно снижен по сравнению с нормой важный печёночный белок, известный как альфа-1 антитрипсин. Люди с дефицитом альфа-1 антитрипсина способны воспроизводить данный белок, однако заболевание препятствует его попаданию в кровоток и способствует его накоплению в печени.
Белок альфа-1 антитрипсин защищает лёгкие от повреждения благодаря естественным ферментам. Когда показатели белка слишком низкие или белок отсутствует, лёгкие могут повреждаться, что ведёт к затруднению функции дыхания и, примерно у 75% людей в подобном состоянии, к эмфиземе - «вздутию» легких. Люди, страдающие данным заболеванием, также находятся в группе риска по развитию цирроза печени.
Каково симптомы недостаточности альфа-1 антитрипсина?
Первыми обычно являются симптомы поражения лёгких, в том числе одышка либо затрудненное дыхание. Необъяснимая потеря массы тела и бочкообразная грудная клетка обычно указывающая на наличие эмфиземы, также являются проявлениями данного заболевания. Поскольку болезнь прогрессирует, могут появиться симптомы, типичные для эмфиземы и цирроза печени, в том числе:
- хронический кашель
- отек лодыжек и стоп
- желтуха
- скопление жидкости в брюшной полости (асцит)
- усталость
Как недостаточносьт альфа-1 антитрипсина диагностируется и лечится?
Физические признаки, такие как бочкообразная грудная клетка и проблемы с дыханием, могут вызвать у вашего врача предположение о недостаточности альфа-1 антитрипсина. Диагноз поможет подтвердить специфическое исследование крови на наличие белка альфа-1 антитрипсина.
Стандартного курса лечения недостаточности альфа-1 антитрипсина нет, но она может быть излечена путём возмещения данного белка в кровотоке. Однако позиция экспертов насчёт того, насколько эффективна эта тактика и к кому её можно применять, до сих пор до конца не ясна. Другими подходами к терапии недостаточности альфа-1 антитрипсина являются лечение осложнений, таких как эмфизема легких и цирроз печени. Они включают в себя лечение антибиотиками для предотвращения респираторных инфекций, ингаляционную лекарственную терапию для облегчения дыхания, диуретики и прочие меры, чтобы избежать любого скопления жидкости в брюшной полости.
Личные привычки, такие как избегание употребления алкоголя, прекращение курения и переход на оздоравливающую диету, также могут помочь предотвратить ухудшение состояния и развитие осложнений. Ваш врач или диетолог может порекомендовать вам наиболее подходящую для вас диету.
Поскольку заболевание влияет на лёгкие, люди с данным заболеванием более подвержены респираторным инфекциям. Вследствие этого рекомендуется вакцинация от гриппа, чтобы предотвратить возникновение данных инфекций и развитие пневмонии. Если вы чувствуете, что у вас развивается насморк или кашель, свяжитесь со своим лечащим врачом, чтобы как можно быстрее приступить к лечению. Временами состояние лёгких или печени продолжает ухудшаться несмотря на лечение. В подобных случаях может быть рекомендована пересадка печени.
Каковы прогнозы для людей с наследственными заболеваниями печени?
При соответствующем лечении недостаточность альфа-1 антитрипсина обычно не ведет к летальному исходу. Однако осложнения, связанные с данными заболеваниями, могут иметь место. Очень важно, чтобы люди с наследственными заболеваниями печени регулярно наблюдались специалистами.
Читайте также: