КТ при синдроме Тричера Коллинза
Добавил пользователь Дмитрий К. Обновлено: 10.01.2026
а) Терминология:
1. Аббревиатура:
• Синдром Тричера Коллинза (СТК)
2. Синонимы:
• Синдром Тричера Коллинза-Франческетти
• Нижнечелюстно-лицевой дизостоз (НЧЛД1)
3. Определение:
• Мальформация лицевого скелета: скошенные вниз глазные щели, микрогнатия, гипоплазия скулового комплекса, микротия, макростомия, колобома
б) Визуализация:
1. Общая характеристика:
• Лучший диагностический критерий:
о Симметричная микрогнатия
о Гипоплазия скулового комплекса
о Двухсторонняя атрезия наружного слухового канала (НСК)
• Локализация:
о Нижняя челюсть, лицо, уши
2. Рекомендации по визуализации:
• Лучший метод визуализации:
о КТ высокого разрешения в костном окне
• Выбор протокола:
о КТ в костном окне верхней и нижней челюсти, височных костей с мультипланарным реформатированием и реконструкциями в объеме
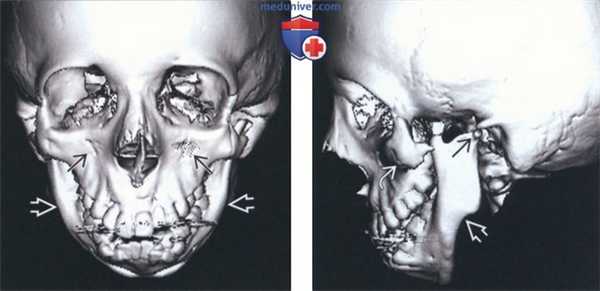
(Слева) На трехмерной реконструкции (КТ, вид спереди) у девочки 13 лет с синдромом Тричера Коллинза (СТК) определяются двухсторонние симметричные впадины на скулах и микрогнатия.
(Справа) На трехмерной реконструкции (КТ, вид сбоку) у этой же пациентки визуализируется маленький мыщелок нижней челюсти. Угол нижней челюсти тупой, видна антегониальная вырезка. Определяется выраженная гипоплазия скулового комплекса и скуловой дуги без слияния со сводом черепа сзади. Также определяется атрезия наружного слухового прохода.
3. КТ при синдроме Тричера Коллинза:
• КТ в костном окне:
о Изменения височных костей:
- Стеноз/атрезия НСК
- Снижение/отсутствие пневматизации сосцевидных отростков
- Гипоплазия/атрезия полости среднего уха
- Мальформация, слияние или отсутствие слуховых косточек
- Стеноз/атрезия овального окна
- Мальформация улитки (уплощение завитков) или норма
- Мальформация горизонтального полукружного канала (или норма)
о Симметричная микрогнатия
о Гипоплазия скулового комплекса
о Гипоплазия мыщелков и венечных отростков
о Выраженная антегониальная вырезка
о Вариабельная расщелина неба
в) Дифференциальная диагностика синдрома Тричера Коллинза:
1. Двухсторонняя фациальная микросомия:
• Окуло-аурикуло-вертебральная дисплазия
2. Преаксиальный акрофациальный дизостоз (синдром Нагера):
• Аналогичен СТК + аномалии конечностей
3. Врожденная дисплазия наружного уха:
• Дисплазия НСК + деформация слуховых косточек
• Отсутствие деформаций нижней или верхней челюсти
4. Бранхио-ото-ренальный синдром:
• Аномалии височной кости, киста жаберной щели, кисты почек
5. Х-сцепленный верхнечелюстнолицевой дизостоз:
• Микрогнатия, срединная гипоплазия лица, аномалии уха и глазных щелей
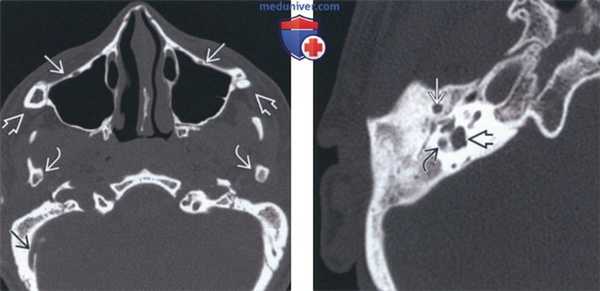
(Слева) На аксиальной КТ в костном окне у этой же пациентки определяется гипоплазия скулового комплекса со скошенностью верхней челюсти кзади, отсутствием скуловых дуг и гипоплазией мыщелков нижней челюсти. Обратите внимание на атрезию наружного слухового канала (НСК), отсутствие пневматизации сосцевидного отростка и расширение эмиссарной вены в сосцевидном отростке.
(Справа) На аксиальной КТ в костном окне у девушки 16 лет с СТК определяется атрезия НСК, отсутствие полости среднего уха и слуховых косточек, вентральное расположение нисходящего канала ЧМН VII, расширение преддверия и мальформация горизонтального полукружного канала.
г) Патология. Общая характеристика:
• Генетика:
о Аутосомно-доминантный тип с различным фенотипом
о Частота: 1/25000-50000 новорожденных
о Локус на генетической карте: 5q32-q33.1
о Мутации генов TCOF1, POLR1С или POLR1D
о Приводят к дефектам нукпеолярного транспортного белка, необходимого при формировании челюстно-лицевой области
д) Клинические особенности:
1. Проявления:
• Типичные признаки/симптомы:
о Скошенные книзу глазные щели (100%)
о Гипоплазия нижней челюсти/скулового комплекса
о Микрогнатия с ретрузией подбородка
о Колобома нижнего века у 75% пациентов
о Микротия, стеноз/атрезия НСК, кондуктивная тугоухость
о Обструкция/гипоплазия дыхательных путей
• Другие признаки/симптомы:
о Расщелина неба в 1/3 случаев
о Задержка моторного/речевого развития
о Гипоплазия или отсутствие околоушных слюнных желез
2. Лечение:
• Респираторная поддержка
• Реконструктивная хирургия
• Слуховые аппараты
• Стимуляция развития
• Ранние интервенционные вмешательства
е) Диагностическая памятка:
1. Следует учесть:
• СТК как причину симметричной микрогнатии, гипоплазии скулового комплекса, аномалий наружного уха
• Изменения, напоминающие СТК+деформации конечностей = синдром Нагера
2. Советы по интерпретации изображений:
• Отсутствие скуловых дуг и скошенность передней стенки верхней челюсти кзади
3. Заключение:
• Ищите окулярную колобому
ж) Список использованной литературы:
1. Ibrahim A et al: Combined soft and skeletal tissue modelling of normal and dysmorphic midface postnatal development. J Craniomaxillofac Surg. 44(11):1777-1785, 2016
2. Plomp RG et al: Treacher Collins syndrome: A systematic review of evidence-based treatment and recommendations. Plast Reconstr Surg. 137(1):191-204, 2016
3. Tse WK: Treacher Collins syndrome: New insights from animal models. Int J Bio-chem Cell Biol. 81 (Pt A):44-47, 2016
4. Robson CD: Congenital hearing impairment. Pediatr Radiol. 36(4):309-24, 2006
5. Teber OA et al: Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 12(11):879-90, 2004
Синдром Тричера-Коллинза ( Мандибулофациальный дизостоз , Синдром Тричера Коллинза-Франческетти , Челюстно-лицевой дизостоз )
Синдром Тричера Коллинза – это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
МКБ-10
Общие сведения
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.
Причины
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная – в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Патогенез
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности – дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
Симптомы
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Осложнения
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагностика
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Прогноз и профилактика
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича – 2009.
2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири – 2007 - №3.
Синдром удлиненного интервала QT
Синдром удлиненного интервала QT – генетически гетерогенное наследственное состояние, характеризующееся нарушением структуры и функциональности некоторых ионных каналов кардиомиоцитов. Выраженность проявлений патологии колеблется в очень широких пределах – от практически бессимптомного течения (выявляются только электрокардиологические признаки) до тяжелой глухоты, обмороков и аритмий. Определение синдрома удлиненного интервала QT производится на основании данных электрокардиологических исследований и молекулярно-генетических анализов. Лечение зависит от формы патологии и может включать в себя постоянный или курсовой прием бета-андреноблокаторов, препаратов магния и калия, а также установку дефибриллятора-кардиовертера.
Синдром удлиненного интервала QT – группа кардиологических расстройств генетической природы, при которых нарушается прохождение ионных токов в кардиомиоцитах, что способно приводить к аритмиям, обморокам и внезапной сердечной смерти. Впервые подобное состояние было выявлено в 1957 году норвежскими врачами А. Джервеллом и Ф. Ланге-Нильсеном, которые описали сочетание у больного врожденной глухоты, синкопальных приступов и удлинения интервала QT. Несколько позже, в 1962-64 годах были выявлены схожие симптомы у пациентов, имеющих нормальный слух – такие случаи были описаны независимо друг от друга К. Романо и О. Уорд.
Это, а также дальнейшие открытия определили разделение синдрома удлиненного интервала QT на два клинических варианта – Романо-Уорда и Джервелла-Ланге-Нильсена. Первый наследуется по аутосомно-доминантному механизму, его частота в популяции составляет 1 случай на 5 000 населения. Встречаемость синдрома удлиненного интервала QT типа Джервелла-Ланге-Нильсена колеблется в пределах 1-6:1 000 000, он характеризуется аутосомно-доминантным путем наследования и более выраженными проявлениями. По некоторым данным, все формы синдрома удлиненного интервала QT ответственны за треть случаев внезапной сердечной смерти и около 20% внезапной младенческой смерти.
Причины и классификация
В настоящее время удалось идентифицировать 12 генов, мутации в которых приводят к развитию синдрома удлиненного интервала QT, все они кодируют те или иные белки, входящие в состав ионных каналов кардиомиоцитов, отвечающих за натриевый или калиевый ионный ток. Удалось также найти причины различий в клиническом течении этого заболевания. Аутосомно-доминантный синдром Романо-Уорда обусловлен мутацией только одного гена и поэтому может протекать бессимптомно или, как минимум, с отсутствием нарушений слуха. При типе Джервелла-Ланге-Нильсена имеется дефект двух генов – этот вариант, помимо кардиологических симптомов, всегда сопровождается двухсторонней нейросенсорной глухотой. На сегодняшний день известно, мутации каких генов обуславливают развитие синдрома удлиненного интервала QT:
- Синдром удлиненного интервала QT тип 1 (LQT1) обусловлен мутацией гена KCNQ1, расположенного на 11-й хромосоме. Дефекты этого гена наиболее часто выявляются при наличии данного заболевания. Он кодирует последовательность альфа-субъединицы одной из разновидностей калиевых каналов кардиомиоцитов (lKs)
- Синдром удлиненного интервала QT тип 2 (LQT2) вызывается дефектами в гене KCNH2, который локализован на 7-й хромосоме и кодирует аминокислотную последовательность белка – альфа-субъединицы другого типа калиевых каналов (lKr).
- Синдром удлиненного интервала QT тип 3 (LQT3) обусловлен мутацией гена SCN5A, расположенного на 3-й хромосоме. В отличие от предыдущих вариантов патологии, при этом нарушается работа натриевых каналов кардиомиоцитов, так как данный ген кодирует последовательность альфа-субъединицы натриевого канала (lNa).
- Синдром удлиненного интервала QT тип 4 (LQT4) – достаточно редкий вариант состояния, вызванный мутацией гена ANK2, который расположен на 4-й хромосоме. Продуктом его экспрессии является белок анкирин В, который в организме человека участвует в стабилизации структуры микротрубочек миоцитов, а также выделяется в клетках нейроглии и сетчатки глаза.
- Синдром удлиненного интервала QT тип 5 (LQT5) – разновидность заболевания, которая обусловлена дефектом в гене KCNE1, локализованном на 21-й хромосоме. Он кодирует один из белков ионных каналов – бета-субъединицу калиевых каналов типа lKs.
- Синдром удлиненного интервала QT типа 6 (LQT6) вызывается мутацией в гене KCNE2, расположенного также на 21-й хромосоме. Продуктом его экспрессии является бета-субъединица калиевых каналов типа lKr.
- Синдром удлиненного интервала QT типа 7 (LQT7, другое название – синдром Андерсена, в честь педиатра Е. Д. Андерсена, описавшего это заболевание в 70-х годах) обусловлен дефектом гена KCNJ2, который локализуется на 17-й хромосоме. Как и в случае предыдущих вариантов патологии, этот ген кодирует одну из белковых цепей калиевых каналов.
- Синдром удлиненного интервала QT типа 8 (LQT8, другое название – синдром Тимоти, в честь К. Тимоти, описавшей это заболевание) вызван мутацией гена CACNA1C, который располагается на 12-й хромосоме. Этот ген кодирует альфа-1-субъединицу кальциевого канала L-типа.
- Синдром удлиненного интервала QT тип 9 (LQT9) обусловлен дефектом гена CAV3, локализованного на 3-й хромосоме. Продуктом его экспрессии является белок кавеолин 3, участвующий в формировании множества структур на поверхности кардиомиоцитов.
- Синдром удлиненного интервала QT тип 10 (LQT10) – причина этой разновидности заболевания кроется в мутации гена SCN4B, который располагается на 11-й хромосоме и отвечает за аминокислотную последовательность бета-субъединицы натриевых каналов.
- Синдром удлиненного интервала QT тип 11 (LQT11) вызывается дефектами в гене AKAP9, расположенном на 7-й хромосоме. Он кодирует специфический белок – А-киназу центросом и комплекса Гольджи. Функции этого протеина на сегодняшний день изучены недостаточно.
- Синдром удлиненного интервала QT тип 12 (LQT12) обусловлен мутацией гена SNTA1, локализованного на 20-й хромосоме. Он кодирует альфа-1-субъединицу белка синтрофина, участвующего в регуляции деятельности натриевых каналов кардиомиоцитов.
Несмотря на широкое генетическое разнообразие синдрома удлиненного интервала QT, общие звенья его патогенеза в целом одинаковы для каждой из форм. Данное заболевание относят к группе каналопатий из-за того, что его причиной выступают нарушения в строении тех или иных ионных каналов. В результате этого процессы реполяризации миокарда происходят неравномерно и не одновременно в различных частях желудочков, что становится причиной удлинения интервала QT. Кроме того, значительно возрастает чувствительность миокарда к влияниям симпатической нервной системы, что становится причиной частых тахиаритмий, способных приводить к жизнеугрожающим фибрилляциям желудочков. При этом у разных генетических типов синдрома удлиненного интервала QT отмечается различная чувствительность к тем или иным воздействиям. Например, LQT1 характеризуется синкопальными приступами и аритмией при физической нагрузке, при LQT2 аналогичные проявления наблюдаются при громких и резких звуках, для LQT3, напротив, более характерно развитие аритмий и фибрилляций в спокойном состоянии (например, во сне).
Симптомы удлиненного интервала QT
Проявления синдрома удлиненного интервала QT достаточно разнообразны. При более тяжелом клиническом типе Джервелла-Ланге-Нильсена у больных отмечается глухота, частые обмороки, головокружения, слабость. Кроме того, в ряде случаев при этом состоянии регистрируются эпилептоподобные судорожные припадки, что нередко приводит к неправильной диагностике и лечению. По данным некоторых врачей-генетиков, от 10 до 25% больных с синдромом удлиненного интервала QT получают неправильное лечение, и у них развивается внезапная сердечная или младенческая смерть. Возникновение тахиаритмий и синкопальных состояний зависит от внешних влияний – например, при LQT1 это может происходить на фоне физической нагрузки, при LQT2 потеря сознания и фибрилляция желудочков может возникать от резких и громких звуков.
Более легкая форма синдрома удлиненного интервала QT (тип Романо-Уорда) характеризуется преходящими синкопальными состояниями (обмороками) и редкими приступами тахиаритмии, однако нарушения слуха при этом отсутствуют. В ряде случаев подобная форма заболевания вообще ничем не проявляет себя, за исключением электрокардиографических данных, и является случайной находкой при медицинском обследовании. Тем не менее, даже при таком течении синдрома удлиненного интервала QT риск внезапной сердечной смерти из-за фибрилляции желудочков во много раз выше, нежели у здорового человека. Поэтому и эта разновидность патологии требует тщательного изучения и профилактического лечения.
Диагностика синдрома удлиненного интервала QT производится на основании изучения анамнеза больного, электрокардиологических и молекулярно-генетических исследований. При расспросе пациента часто обнаруживаются эпизоды обмороков, головокружений, ощущения сердцебиений, но при легких формах патологии их может и не быть. Иногда аналогичные проявления встречаются у кого-либо из родственников пациента, что указывает на семейный характер заболевания.
При любой форме синдрома удлиненного интервала QT будут выявляться изменения на ЭКГ – увеличение интервала QT до 0,6 секунд и более, возможно увеличение амплитуды зубца Т. Сочетание таких ЭКГ-признаков с врожденной глухотой говорит о наличии синдрома Джервелла-Ланге-Нильсена. Кроме того, часто необходимо холтеровское мониторирование работы сердца на протяжении суток для выявления возможных приступов тахиаритмий. Определение синдрома удлиненного интервала QT при помощи методов современной генетики на сегодняшний день возможно в отношении практических всех генетических типов этого заболевания.
Лечение синдрома удлиненного интервала QT
Терапия синдрома удлиненного интервала QT достаточно сложна, многие специалисты рекомендуют при этом заболевании одни схемы и отвергают другие, но какого-либо единого протокола лечения этой патологии не существует. Универсальными препаратами считаются бета-адреноблокаторы, которые уменьшают риск развития тахиаритмий и фибрилляций, а также снижают степень симпатических воздействий на миокард, но при LQT3 они малоэффективны. В случае синдрома удлиненного интервала QT типа 3 более разумно использовать антиаритмические препараты класса В1. Эти особенности лечения заболевания повышают потребность в молекулярно-генетической диагностике для определения типа патологии. В случае частых приступов тахиаритмий и высокого риска развития фибрилляции рекомендуется имплантация кардиостимулятора или дефибриллятора-кардиовертера.
Прогноз
Прогноз синдрома удлиненного интервала QT, по мнению большинства специалистов, неопределенный, так как это заболевание характеризуется широким спектром выраженности симптомов. Кроме того, отсутствие проявлений патологии, за исключением электрокардиографических данных, не гарантирует внезапного развития фатальной фибрилляции желудочков под воздействием внешних или внутренних факторов. При выявлении синдрома удлиненного интервала QT необходимо произвести тщательное кардиологическое обследование и генетическое определение типа заболевания. На основе полученных данных разрабатывается схема лечения, призванная снизить вероятность внезапной сердечной смерти, или принимается решение об имплантации кардиостимулятора.
Синдром Паллистера-Киллиана ( Синдром мозаичной изохромосомы 12р , Тетрасомия 12р )
Синдром Паллистера-Киллиана — это крайне редкое генетическое заболевание, которое возникает при тетрасомии по 12-й хромосоме. Его развитие связывают с Х-рецессивным типом наследования. Состояние проявляется множественными патологиями перинатального периода, тяжелыми формами умственной отсталости, разнообразными врожденными аномалиями внутренних органов и стигмами дизэмбриогенеза. Диагностика синдрома Паллистера включает генетическое тестирование, инструментальную визуализацию (УЗИ, КТ, МРТ), неврологическое обследование. Пациентам назначается поддерживающее лечение, комплексная программа реабилитации, регулярное диспансерное наблюдение.
Синдром Паллистера (Паллистера-Киллиана) имеет синонимичные названия тетрасомия 12р, синдром мозаичной изохромосомы 12р. Он встречается крайне редко, в медицинской литературе есть данные о 30 случаях болезни, причем 6 пациентов проживают в Российской Федерации. Патология впервые описана в 1977 г. ученым Ф. Паллистером. В 1981 г. М. Теслер-Никола и В. Киллиан независимо друг от друга более подробно охарактеризовали клиническую картину синдрома. Первый пренатально диагностированный случай заболевания датируется 1985 г.
Болезнь возникает вследствие редкого мозаичного хромосомного расстройства, при котором в генетическом материале клетки образуются 4 копии короткого плеча 12 хромосомы вместо двух, которые должны присутствовать в норме. Эти дополнительные копии, как правило, расположены в одной из хромосом (изохромосома). Предполагается, что у синдрома Паллистера Х-сцепленный рецессивный тип наследования, но данных для подтверждения этой гипотезы недостаточно.
Заболевание появляется в результате генных нарушений, которые вызваны присутствием мозаичного клона mos 47,+ i(12)(p10)/46. Мутация формируется при нерасхождении сестринских хроматид в фазе мейоза II при образовании женских половых клеток. После оплодотворения такой яйцеклетки наблюдается постзиготическое ошибочное деление центромеры и частичная ее потеря при последующих митозах, поэтому только часть клеток тела ребенка имеют тетрасомию 12р.
Синдром Паллистера — тяжелая болезнь, которая может завершаться внутриутробной гибелью плода. В остальных случаях в анамнезе определяются патологии течения беременности в виде раннего токсикоза, угрозы прерывания на поздних сроках, задержки внутриутробного развития плода. У новорожденных отмечаются неврологические осложнения вследствие гипоксически-ишемической энцефалопатии, проблемы с самостоятельным дыханием, средняя или низкая оценка по шкале Апгар.
Больные с синдромом Паллистера имеют специфические особенности лицевого скелета: выступающий лоб с завышенной линией роста волос, низко посаженные деформированные уши, широкое расстояние между глазами. Также встречаются маленький вздернутый нос, вывернутые ноздри, большое расстояние от нижнего края ноздрей до верхней губы. На коже тела выявляются очаги депигментации.
К характерным особенностям синдрома относят диспропорциональное развитие конечностей с укорочением их проксимальных отделов, что сопровождается контрактурами суставов, стойким ограничением объема активных движений. Часто беспокоят проблемы со слухом и зрением. Для синдрома Паллистера-Киллиана типичны пороки сердца (дефект межжелудочковой перегородки, коарктация аорты, открытый Боталлов проток), диафрагмальная грыжа.
При тетрасомии 12р отмечается задержка психомоторного развития, умственная отсталость. Дети поздно начинают переворачиваться, сидеть и ползать, зачастую они не говорят даже в возрасте 1-3 лет, а только произносят отдельные звуки (гуление) или слоги. Наблюдается отставание в росте и физическом развитии, пациенты плохо набирают массу тела, имеют астеническое телосложение. Неврологические нарушения проявляются эпилепсией, мышечной гипотонией, тремором.
У страдающих болезнью Паллистера выявляются множественные пороки на фоне тяжелого неврологического дефицита, что обуславливает раннюю инвалидизацию таких пациентов. Осложнения синдрома связаны с задержкой речи и психического развития, из-за чего больным нужен постоянный уход, квалифицированная медицинская помощь. Неблагоприятный исход обычно наблюдается при тяжелых врожденных пороках, которые с трудом поддаются хирургической коррекции.
Своевременная постановка диагноза затруднена, что обусловлено большой редкостью синдрома Паллистера, наличием тканеспецифичного мозаицизма, что создает сложности при генетических анализах. Перед посещением генетика такие дети зачастую проходят осмотр у большого круга специалистов: педиатров, неврологов, кардиологов, эндокринологов. Для диагностики синдрома Паллистера назначаются следующие методы:
- FISH-анализ. Такой метод генетического тестирования является наиболее точным для определения тетрасомии 12р. Для исследования используются клетки буккального эпителия или культура фибробластов, среди которых до 50-65% имеют характерную мутацию. При анализе лимфоцитов только у 2% клеток обнаруживается дополнительное короткое плечо 12-й хромосомы.
- Консультация невролога. Больным рекомендован детальный неврологический осмотр с оценкой рефлекторной возбудимости, мышечного тонуса, уровня развития психомоторных функций. Обязательно проверяются органы чувств, поскольку у таких детей часто возникают тугоухость, снижение зрения.
- Инструментальные методы. Для выявления сопутствующих врожденных пороков выполняется комплексное обследование с применением УЗИ органов брюшной полости, рентгенографии грудной клетки, эхокардиографии. По показаниям проводится нейросонография, КТ или МРТ головного мозга.
Лечение синдрома Паллистера
Консервативная терапия
Специальные методы лечения синдрома Паллистера пока не разработаны. Основу терапии составляют поддерживающие мероприятия, которые уменьшают клинические проявления болезни, стимулируют формирование отсутствующих психомоторных навыков, помогают адаптировать больного к жизни с инвалидностью. План терапии синдрома включает несколько направлений:
- Метаболические препараты. Лекарства, стимулирующие энергетический обмен, а также витаминно-минеральные комплексы и гомеопатические средства назначаются для поддержки работы миокарда, улучшения функционирования нервной системы.
- Реабилитация. Пациентам показана комплексная реабилитационная программа с участием инструкторов ЛФК, специалистов по механотерапии, дефектологов. Такие мероприятия направлены на развитие у детей устной речи, коммуникативных навыков, улучшения моторных функций.
Оперативное лечение
Сердечные пороки, встречающиеся при синдроме, требуют оперативной коррекции, которая в основном проводится в первый год жизни ребенка. Сроки оказания кардиохирургической помощи определяются тяжестью нарушений кровообращения.
С учетом отсутствия этиопатогенетических методов терапии синдрома Паллистера, тяжести клинической симптоматики и формирования инвалидности в раннем возрасте, прогноз неблагоприятный. Поскольку цель проводимых лечебных мероприятий — улучшение качества жизни пациентов, при их успешной реализации удается значительно развить психомоторные навыки. Специфическая профилактика синдрома отсутствует.
1. Реабилитация больных с синдромом Паллистера-Киллиана с учетом особенностей их метаболизма — клинические наблюдения/ Ю.Б. Гречанина, И.А. Максютина, М.Б. Грузкова// Клиническая генетика и перинатальная диагностика. — 2018. — №1(4).
2. Клинический случай синдрома Паллистера-Киллиана у ребенка/ Т.В. Судовская, Н.Ш. Кокоева, Ю.А. Бобровская, А.А. Макарова// Российская педиатрическая офтальмология. — 2017. — №3.
3. Синдром Паллистера-Киллиана: особенности пре- и перинатальной диагностики/ Н.В. Шилова, Ю.О. Козлова, Н.А. Демина, М.С. Петухова// Медицинская генетика. — 2012. — №11(3).
4. Синдром Паллистера-Киллиана: сонографическая, молекулярно-цитогенетическая и патоморфологическая диагностика/ А.Д. Политыко, И.В. Новикова, Э.И. Мараховская, В.Н. Кошкина// Медицинская генетика. — 2009. — №8.
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
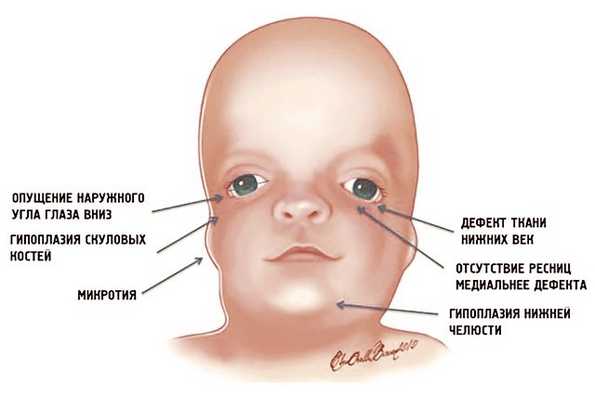
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
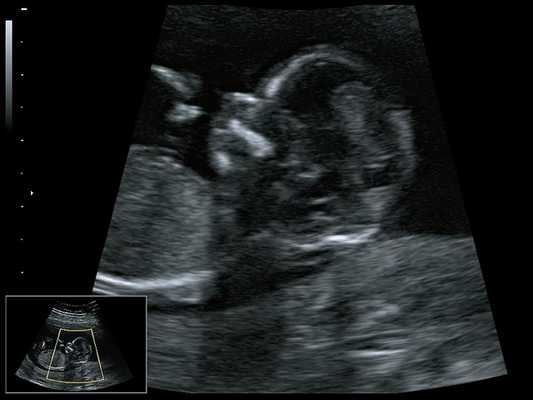
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Читайте также:
- Лечение герминогенной опухоли яичника у беременной - тактика
- Симптомы поражения ипритом. Особенности ипритных поражений
- Вывихи в пястно-фаланговых суставах. Диагностика, лечение
- Развитие фолликула. Последовательность развития яйцеклетки
- Микроорганизмы. Типы микроорганизмов. Классификация микроорганизмов. Прионы.