Мальформация коренного зуба (синдром Жубера) на МРТ
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
УЗИ, МРТ при синдроме Жубера у плода
а) Определения:
• СЖ и родственные заболевания:
о Группа генетически обусловленных заболеваний, имеющих аутосомно-рецессивный тип наследования и характеризующихся цилиарной дисфункцией
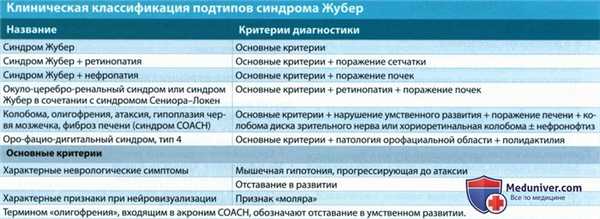
о Шесть подтипов; для каждого характерно наличие признака «моляра»:
- В научной литературе ведутся споры, касающиеся классификации заболеваний, поскольку фенотипические проявления широко варьируют
• Критерии диагностики классической формы СЖ:
о Аномалия развития ромбовидного мозга, проявляющаяся признаком «моляра» при МРТ
о Снижение интеллекта о Мышечная гипотония
• СЖ является представителем цилиопатий:
о Группа наследственных дефектов первичных (неподвижных) ресничек, обусловливающих широкий спектр схожих синдромов с поражением печени, почек и многих систем органов
о Первичные реснички играют важную роль в эмбриогенезе, и с их дефектами связано возникновение многих наследственных заболеваний
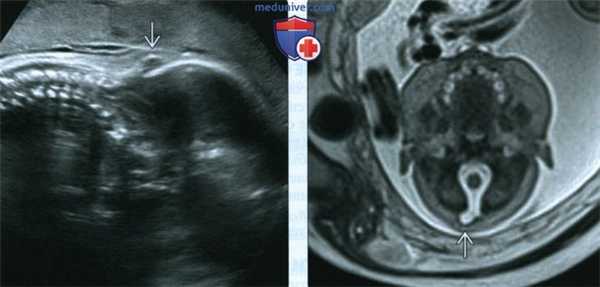
(Слева) УЗИ шейного отдела позвоночника в сагиттальной плоскости. Обнаружено затылочноеэнцефалоцеле небольших размеров Нарушение не было задокументировано во время данного исследования - энцефалоцеле было позднее обнаружено на МРТ плода, что заставило вернуться к ранее выполненным УЗИ и ретроспективно подтвердить находку.
(Справа) Тот же случай. МРТ основания черепа плода, Т2-ВИ, поперечный срез. Отчетливо визуализируется затылочное энцефалоцеле. Данная находка характерна для СЖ, однако часто упускается из внимания во время УЗИ, особенно если нарушение впервые появилось в III триместре.
б) Лучевая диагностика:
1. УЗИ при синдроме Жубера у плода:
• Увеличение ТВП: неспецифический признак, свидетельствующий о возможном повышении риска заболевания
• Аномалии структур ЗЧЯ: расщепление мозжечка, дисгене-зия или ротация червя мозжечка
• Изменения IV желудочка:
о Поперечная плоскость: верхняя часть моста в форме крыла летучей мыши; переднезадний размер больше поперечного
о Сагиттальная плоскость: крыша желудочка вытянута, округлая; заостренное ядро шатра мозжечка не определяется
• Может визуализироваться затылочное энцефалоцеле небольших размеров, а также изменения мозолистого тела
• Характер дыхания плода: возможно периодическое гипер-пноэ (140-160 дыхательных движений в минуту)
2. МРТ при синдроме Жубера у плода:
• Признак «моляра» (определяется не только на поперечном срезе МРТ, но и при УЗИ):
о Глубокая межножковая ямка:
- Дисгенезия перешейка ствола мозга → удлинение мостосреднемозгового соединения
о Утолщенные, прямые, вытянутые верхние ножки мозжечка
о Гипоплазия червя, возможна его ротация
• Дно IV желудочка выгнуто кпереди из-за недостаточного пересечения ножек мозжечка на уровне покрышки:
о Определяется только на поперечном срезе на уровне верхней части моста
• Расщепление мозжечка по средней линии
• Расширение ретроцеребеллярного пространства
• У пораженных плодов определяется выраженное отклонение указанных характеристик от нормы:
о Мостосреднемозговой угол
о Отношение переднезадних размеров межножковой ямки и среднего мозга/перешейка
о Отношение переднезаднего размера IV желудочка к поперечному
• Патологические находки в супратенториальном пространстве: отсутствие ППП, изменения мозолистого тела, кортикальная дисплазия
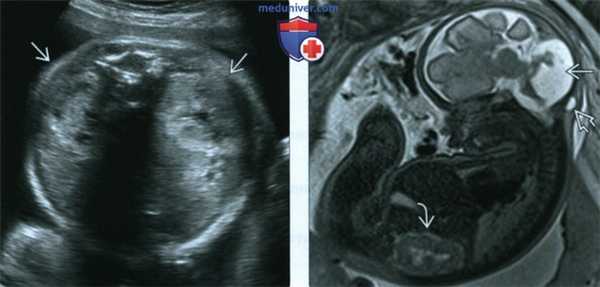
(Слева) Тот же плод. УЗИ брюшной полости. Обе почки увеличены и гипер-эхогенны, что позволяет заподозрить СЖ с поражением почек.
(Справа) МРТ в сагиттальной плоскости. Отмечаются аномалии структур ЗЧЯ, затылочное энцефалоцеле и увеличение почки. МРТ плода позволяет прицельно исследовать определенную часть тела (в данном случае - головной мозг), однако нередко возможно получить достаточный объем диагностических сведений на основании обычных «поисковых» методов.
в) Дифференциальная диагностика синдрома Жубера у плода. Другие пороки развития структур ЗЧЯ:
• Мальформация Денди-Уокера:
о Полость IV желудочка сообщается с большой цистерной
о Отсутствует признак «моляра»
о Средний мозг нормальных размеров, при СЖ он тоньше
• Мальформация Арнольда-Киари:
о Облитерация большой цистерны
о Грыжевое выпячивание миндалин мозжечка, придающее мозжечку форму банана
о Обычно сочетается с дефектом заращения нервной трубки
• РБЦ:
о Глубина большой цистерны >10 мм
о Без сопутствующих структурных аномалий
г) Патологоанатомические особенности:
1. Общие сведения:
• Генетические факторы:
о Вариабельность фенотипа, предположительно, обусловлена олигогенной моделью наследования:
- Фенотип формируется под влиянием конкурентных эффектов >2 отдельных генов
о Аутосомно-рецессивный тип наследования (к 2014 г. установлены мутации 21 гена):
- INPP5E, АНН, NPHP1, СЕР290, TMEM67/MKS3, RPGR1P1L, ARL13B, CC2D2A, INPP5E, KIF7, OFD1, TCTN1, TCTN2, ТМЕМ2167
- Франкоканадцы в Квебеке являются носителями множественных мутаций с эффектом основателя (C5orf42, CC2D2A и ТМЕМ231)
- 1% евреев-ашкеназов являются носителями мутации с эффектом основателя - p.R73L гена ТМЕМ216
- В общине гуттеритов частота носительства мутации с эффектом основателя - p.R18X гена ТМЕМ237 - составляет 5,8%
о Описаны формы, имеющие Х-сцепленный тип наследования
• Сопутствующая патология:
о За развитие большинства нарушений ответственны дефекты структуры ± функции первичных ресничек:
- Фиброз печени, кистозная болезнь почек
- Менингоэнцефалоцеле
- Патология органа зрения (колобома, хориоретинальная дисплазия)
- Полидактилия
- Расщелина нёба, опухоли языка
- Лицевая дизморфия
- ВПС (редко)
2. Макроскопические изменения и исследование операционного материала:
• Изменения базальных ядер и среднего мозга
• Измененные волокна ножек мозжечка
• Уменьшение объема ядер глазодвигательного нерва
• Гипо- или аплазия червя мозжечка
д) Клинические особенности:
1. Клиническая картина:
• У плода:
о В большинстве случаев заболевание впервые проявляется патологией структур ЗЧЯ ± полидактилией по результатам скринингового УЗИ:
- Порок развития ромбовидного мозга, проявляющийся признаком «моляра»
- Затылочное энцефалоцеле
• В постнатальном периоде:
о Широкий спектр других фенотипических проявлений
о Дополнительные, но не обязательные признаки - нерегулярное дыхание, нарушение движений глазных яблок
о Характерные черты лица: высокие округлые брови, ноздри, вывернутые кпереди, рот треугольной формы, низкорасположенные уши
2. Демографические особенности:
• Заболеваемость СЖ в США составляет 1:80 000-100 000
• Точная распространенность заболевания неизвестна; предположительно, большинство случаев остаются недиагно-стированными
3. Естественное течение и прогноз:
• Исход не зависит от выраженности патологических находок, обнаруженных методами лучевой диагностики
• Большинство детей доживают до взрослого возраста
• У пораженных детей присутствует множество нарушений:
о Задержка в развитии, умственная отсталость:
- Средний возраст начала самостоятельного сидения -19 мес.
- Средний возраст начала хождения среди детей, способных к обучению, — 4 года
- Атаксия обусловлена пороком развития ромбовидного мозга
о Глазодвигательная апраксия, артикуляционная диспраксия:
- Артикуляция страдает сильнее, чем восприятие
о Мышечная гипотония
о Различные нарушения со стороны дыхательной системы:
- Гиперпноэ и апноэ (внезапная смерть новорожденных чаще всего связана с приступами апноэ)
о Судорожный синдром чаще всего наблюдают при наличии сопутствующих пороков ЦНС:
- Нарушения, устойчивые к лечению, связаны с низкой долгосрочной выживаемостью
• Поведенческие нарушения (импульсивность, персеверация, приступы гнева) у детей более старшего возраста
• Риск повторного возникновения - 25%; клиническая картина у сиблингов широко варьирует
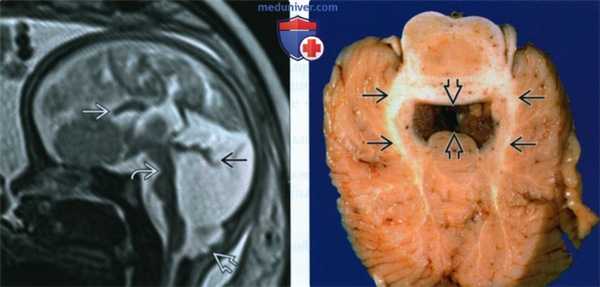
(Слева) МРТ, Т2-ВИ, сагиттальная плоскость. Представлены характерные признаки СЖ: дисгенезия мозолистого тела тонкий перешеек ствола мозга, патологическая ротация червя мозжечка э и затылочное энцефалоцеле.
(Справа) Аутопсия плода с СЖ. Строение головного мозга на срезе соответствует признаку «моляра». Верхние ножки мозжечка прямые и утолщенные («корни зуба»), очертания IV желудочка изменены и по форме напоминают «крыло летучей мыши».
4. Лечение синдрома Жубера у плода:
• Предлагают выполнение биопсии ворсин хориона или амниоцентеза, особенно при беременности высокого риска:
о В случае положительного результата предлагают прерывание беременности
• Живорожденный младенец:
о При подтверждении диагноза СЖ требуется тщательное обследование ребенка на наличие возможных осложнений
о Мутации отдельных генов → очень высокая вероятность специфических осложнений:
- В результате мутаций генов NPHP1, RPGRIP1L, СЕР290 возникает нефронофтиз:
Для контроля функции почек и составления плана лечения требуется наблюдение у нефролога
- Мутации гена ТМЕМ67 практически в 100% случаев приводят к появлению врожденного фиброза печени
• Последующие беременности:
о УЗИ на ранних сроках, МРТ в 20-22 нед.
о При наличии установленной мутации возможна пренатальная диагностика с помощью исследования ДНК:
- В 50% случаев конкретные мутации неизвестны
е) Особенности диагностики. Важно знать:
• Для уточнения аномалий ЦНС, в особенности структур ЗЧЯ, выполняют МРТ плода:
о Наиболее затруднена дифференциальная диагностика с мальформацией Денди-Уокера
о Точная диагностика имеет важное значение, поскольку прогноз и риск повторного возникновения для данных заболеваний различаются
• Новорожденным показано тщательное обследование и наблюдение за развитием возможных осложнений
ж) Список использованной литературы:
1. Quarello Е et al: Prenatal abnormal features of the fourth ventricle in Joubert syndrome and related disorders. Ultrasound Obstet Gynecol. 43(2):227-32, 2014
2. Valente EM et al: Clinical utility gene card for: Joubert syndrome -update 2013. Eur J Hum Genet. 21(10), 2013
3. Incecik F et al: Joubert syndrome: report of 11 cases. Turk J Pediatr. 54(6):605—11, 2012
4. Poretti A et al: Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol. 32(8): 1459-63, 2011
5. Poretti A et al: Tecto-cerebellar dysraphism with occipital encephalocele: not a distinct disorder, but part of the Joubert syndrome spectrum? Neuropediatrics. 42(4): 170-4, 2011
- Вернуться в оглавление раздела "Акушерство."
Редактор: Искандер Милевски. Дата обновления публикации: 18.11.2021
Мальформация коренного зуба (синдром Жубера) на МРТ
Заболевания
Синдром Жубера (Joubert syndrome) – редкое генетически гетерогенное наследственное заболевание, характеризующееся нарушением формирования мозжечка и структур мозгового ствола с развитием соответствующей неврологической симптоматики. Симптомы данной патологии проявляют значительную вариабельность по своей выраженности, наиболее часто наблюдаются расстройства дыхания, глазодвигательные нарушения и мышечная слабость, возможны нарушения слуха и отставание в интеллектуальном развитии. Диагностика синдрома Жубера производится на основании неврологического осмотра больного, магнитно-резонансной томографии головного мозга, молекулярно-генетических исследований. Специфического лечения на сегодняшний момент не существует, применяют поддерживающие и симптоматические мероприятия.
История
В 1969 году врач ординатор детской больницы Монреаля Мари Жубер с соавторами описала 4 сибсов от состоящих в кровном родстве родителей. Ее первым пациентом был мальчик, госпитализированный в клинику в возрасте 6 мес. До 2002 года этот пациент оставался жив. Название «Синдром Жубер» было предложено Ойгеном Болтшаузером и Вернером Ислером в 1977 году.
Диагностические критерии синдрома Жубер продолжают меняться, но большинство авторов считают обязательным нейрорадиологическим признаком симптом «коренного зуба».
Диагноз «классического» синдрома Жубер основан на наличии трех основных критериев:
- симптом «коренного зуба»: на МРТ в аксиальной плоскости через точку соединения среднего мозга и моста (область перешейка) выявляются признаки гипоплазии червя мозжечка и сопутствующие аномалии ствола мозга. Признак «коренного зуба» включает в себя аномально глубокую межножковую ямку, заметные, прямые и утолщенные верхние ножки мозжечка и гипоплазию червя мозжечка;
- гипотония в раннем детском возрасте с последующим развитием
- атаксии; задержка развития/умственная отсталость.
Дополнительные признаки, часто наблюдаемые у пациентов с синдромом Жубер:
- патологический дыхательный паттерн (чередование тахипноэ и/или апноэ);
- аномальные движения глаз: как правило, окуломоторная апраксия или затруднения при плавном слежении за предметами и подергивание при пристальном взгляде.
Термин «синдром Жубера и связанные расстройства» относится к пациентам с синдромом Жубера, имеющим дополнительные признаки, включая дистрофию сетчатки, заболевания почек, колобомы, затылочное энцефалоцеле, фиброз печени, полидактилию, гамартомы полости рта и другие нарушения. У значительной части пациентов с диагнозом «классический синдром Жубера» с течением времени проявляются дополнительные признаки, представляющие собой «синдром Жубера и связанные расстройства». Учитывая общность главных диагностических критериев.
Этиология
Известны 18 генов, мутации в которых вызывают синдром Жубер: NPHP1, AHI1, CEP290 (NPHP6), TMEM67 (MKS3), RPGRIP1L, CC2D2A, ARL13B, INPP5E, OFD1, TMEM216, KIF7, TCTN1, TCTN2, TMEM237, CEP41, TMEM138, C5orf42, и TTC21B. Вероятно, могут быть вовлечены и другие локусы: примерно у 50% людей с синдромом Жубер и связанными расстройствами была идентифицирована мутация в одном из известных генов; фенотип синдрома Жубер во многих семьях не был связан ни с одним из генов, идентифициро ванных к настоящему времени.
Распространенность синдрома Жубер не определена. Многие авторы приводят цифры от 1:80000 до 1:100000, но эта оценка может быть занижена. Дифференциальную диагностику синдрома Жубер проводят со следующими заболеваниями: нефронофтиз, синдром Когана, врожденный амавроз Лебера, синдром Барде-Бидля, синдром Меккеля, синдром MORM (умственная отсталость, ожирение, дистрофия сетчатки, микропенис), оро-фацио-дигитальный синдром 1-го типа, гидролетальный синдром, акрокаллезный синдром.
Клиническая картина.
Синдром Жубера характеризуется тремя основными признаками: характерной аномалией мозжечка и ствола головного мозга, называемой симптом «коренного зуба», гипотонией и задержкой развития. Часто эти проявления сопровождаются эпизодическим тахипноэ или апноэ и/или атипичными движениями глаз. Нарушения дыхания с возрастом проходят, со временем развивается стволовая атаксия и задержка моторного развития. Кроме того, у пациентов с синдромом Жубера могут наблюдаться дистрофия сетчатки, заболевания почек, колобомы, затылочное энцефалоцеле, фиброз печени, полидактилия, гамартомы полости рта и патология эндокринной системы. Когнитивные способности варьируют от тяжелого дефицита до практически нормального интеллекта. При синдроме Жубера наблюдаются внутри- и межсемейные фенотипические различия.
Пациентам с синдромом Жубера рекомендуется ежегодное наблюдение у ряда специалистов:
- наблюдение педиатра и невролога с оценкой физического, психомоторного и полового развития, дыхательной системы (включая симптомы апноэ);
- консультация нейропсихолога;
- наблюдение офтальмолога (определение остроты зрения, способности следить за предметами, предупреждение развития дистрофии сетчатки);
- УЗИ органов брюшной полости;
- исследование функции печени, включая определение уровня трансаминаз, альбумина, билирубина и протромбинового времени;
- оценка функции почек: измерение артериального давления, определение уровня азота мочевины в крови, концентрации креатинина в сыворотке, клинический анализ крови и анализ мочи с определением концентрационной функции почек.
По рекомендации лечащего врача, в зависимости от клинических проявлений у конкретного пациента, в этот список могут быть включены дополнительные обследования.
Нейрорадиологические особенности
Признак летучей мыши
На сагиттальных срезах почти всегда отмечается отсутствие червя мозжечка, хотя верхняя его часть (разделенная срединной расщелиной) в ряде случаев присутствует. Своеобразная парциальная гипогенезия червя приводит к появлению специфических формообразующих феноменов IV желудочка: его нижняя часть приобретает треугольноподобную форму, а верхняя благодаря частичной сохранности расщепленного червя — форму «летучей мыши». В нижних отделах задней черепной ямки — ниже гипопластичного червя церебеллярные гемисферы примыкают друг к другу по срединной линии. Верхние мозжечковые ножки не пересекаются в задних отделах среднего мозга и могут быть легко идентифицированы на фоне окружающей их спинномозговой жидкости, будучи позиционированы между средним мозгом и мозжечком. Мост уменьшен в переднезаднем размере (возможно, ввиду отсутствия перекреста пирамид).
Признак коренного зуба
Синдром Денди-Уокера: понятие, причины, формы, лечение, прогноз
Аномалии формирования центральной нервной системы — отнюдь не редкость. Эта патология занимает второе место по частоте после врожденных дефектов развития сердца и сосудов и в большинстве случаев представлена гидроцефалией самого разного происхождения. Помимо тяжелых нарушений, сопровождающих патологическое развития мозга, пороки ЦНС несут высокий риск смертельного исхода, а по данным статистики они лидируют по числу смертей в младенческом возрасте.
Синдром Денди-Уокера — одна из разновидностей нарушения формирования головного мозга еще во время внутриутробного развития. И хотя частота его относительно невелика (всего 1 случай на 25-30 тысяч младенцев), диагностируется порок едва ли не у каждого десятого малыша с врожденной гидроцефалией, которая и служит одним из основных проявлений патологии.
Синдром Денди-Уокера — это порок задней черепной ямки, при этом основные структурные изменения касаются мозжечка, ликвороотводящих путей, четвертого желудочка мозга. Аномалия диагностируется во время беременности посредством ультразвукового осмотра, после чего женщине может быть предложено прерывание беременности по медицинским показаниям.
Конечно, любые отклонения в развитии плода — это всегда большой стресс и переживания для родителей, однако в случае врожденного порока мозга надеяться на чудо не приходится — прогноз серьезный, а смертность высока. Малыши с сочетанными пороками мозга и других органов погибают в раннем возрасте как от мозговой дисфункции, так и от присоединившейся инфекции.
Аномалия Денди-Уокера часто сочетается с другими нарушениями и генетическими заболеваниями, зачастую несовместимыми с жизнью — микроцефалия (недоразвитие полушарий мозга), мозговые грыжи. У младенца может быть диагностирован генетически обусловленный поликистоз почек, недоразвитие зрительных нервов и глазных яблок со слепотой, аномалии сердечно-сосудистой системы.
Все эти неблагоприятные факторы, возможность сочетанной патологии многих органов делают синдром Денди-Уокера серьезнейшей проблемой в случае, если малышу дадут возможность родиться. Лечение патологии, как правило, симптоматическое, направленное на поддержание главных систем жизнеобеспечения и борьбу с инфекционными осложнениями. В редких случаях применяют хирургическую операцию, которая лишь облегчает явления гидроцефалии, но не ликвидирует ее полностью.
Почему возникает синдром Денди-Уокера?
Причины аномалий развития задней черепной ямки до сих пор не выяснены, однако выделен ряд факторов, способствующих подобным врожденным порокам:
- Инфицирование во время беременности цитомегаловирусом, перенесенная краснуха;
- Употребление алкоголя, курение, наркомания во время беременности;
- Экстрагенитальная патология, особенно — сахарный диабет у будущей мамы.
Под действием перечисленных причин или среди полного благополучия может возникнуть спонтанная мутация в генах, предрасполагающая к нарушению развития мозга. Особенно высок риск пороков при действии неблагоприятных факторов во время первого триместра гестации, когда и происходит закладка основных структур центральной нервной системы.
В части случаев синдром Денди-Уокера носит наследственно-обусловленный характер, то есть возникает из-за дефекта генов и может передаваться по наследству как рецессивный признак. Если первая беременность протекала с формированием этой патологии, то риск повторного порока в последующем возрастает до 25%.
Что происходит с мозгом при синдроме Денди-Уокера?
Анатомически классический вариант мальформации задней черепной ямки включает:
- Гидроцефальный синдром разной степени выраженности;
- Кистозную полость в задней части черепа с расширенным четвертым желудочком мозга;
- Отсутствие или недоразвитие червя мозжечка, недоразвитие его полушарий.
Червь мозжечка — это структура, расположенная между его половинами и несущая в себе проводящие нервные волокна. При аномалии Денди-Уокера он может быть представлен небольшой щелью или широким пространством между гемисферами органа. При неполном отсутствии червя щелевидное расширение образуется лишь в нижней его части. На фоне патологии этого отдела наблюдается недостаточное развитие и мозжечковых полушарий.
Именно дефект мозжечкового червя в виде расщелины считается характерным признаком аномалии Денди-Уокера, позволяющим отличать ее от недоразвития на фоне других пороков мозга.
Киста четвертого мозгового желудочка может самопроизвольно вскрыться в 3-ий желудочек или субарахноидальное пространство. В этом случае симптомы окклюзии ликворных путей несколько ослабнут. Выраженность гидроцефального синдрома вариабельна — от небольшого расширения желудочковой системы до высокой степени окклюзионной гидроцефалии с отсутствием возможности для циркуляции ликвора.
Многие специалисты отмечают, что у большинства малышей с синдромом Денди-Уокера при рождении гидроцефалии как таковой нет, а формируется она и прогрессивно нарастает в течение первых нескольких месяцев жизни, поэтому факт отсутствия гидроцефального синдрома сразу после родов при наличии диагностированной внутриутробно патологии не является поводом для пересмотра диагноза и необоснованных надежд, с этим связанных.
Более, чем в половине случаев синдрома Денди-Уокера у детей помимо описанных структурных аномалий обнаруживаются и другие дефекты мозга — недоразвитие или отсутствие мозолистого тела, мозговые кисты, недоразвитие или отсутствие извилин, смещения серого вещества относительно его правильной локализации, что еще больше усугубляет течение и без того тяжелой патологии.
По данным МРТ было выделено несколько разновидностей синдрома Денди-Уокера:
- Классический тип аномалии — задняя черепная ямка расширена, четвертый желудочек кистозно изменен, червь мозжечка частично или полностью недоразвит, полушария его гипоплазированы, а намет находится выше, чем в норме, желудочковая система не сообщается с подпаутинным пространством, часто наблюдаются мозговые кисты и отсутствие мозолистого тела, практически у всех пациентов есть гидроцефалия, возможно сдавление стволовых структур. Порок проявляется клинически уже с рождения и имеет неблагоприятный прогноз.
- Вариант Денди-Уокера — морфологические признаки выражены меньше, чем при классической форме, гипоплазирован нижний отдел червя мозжечка, желудочки сообщаются с кистой и ликворными пространствами, обеспечивая отток ликвора, поэтому гидроцефалия наблюдается редко. Задняя черепная ямка имеет нормальные размеры, стволовые структуры не сдавливаются.
- Киста кармана Блейка — расширение желудочковой системы с гидроцефальным синдромом, киста расположена под или за мозжечком, червь развит относительно хорошо. Четвертый желудочек расширен, но не сообщается с затылочной ликворной цистерной.
- Mega cisterna magna — вариант очагового расширения подпаутинного пространства в задней и нижней частях задней ямки черепа с увеличением объема затылочной цистерны, которая сообщается с четвертым желудочком и субарахноидальным пространством.
Проявления заболевания
Симптоматика синдрома Денди-Уокера разнообразна. Возможны как практически нормальное развитие ребенка после рождения, так и грубые неврологические изменения, влекущие тяжелую инвалидность и даже смерть. По некоторым данным, нормальное развитие интеллекта бывает в половине случаев изолированного порока, возможно даже случайное обнаружение синдрома при обследовании взрослых.
дети с синдромом Денди-Уокера
Внутриутробное течение патологии определяется степенью поражения мозга, нарастанием гидроцефалии, наличием других пороков развития. Прогноз значительно хуже при диагностике синдрома до рождения. При глубоких нарушениях в формировании мозга на первый план среди других проявлений выступает гидроцефальный синдром:
- Увеличение диаметра головы;
- Выбухание родничка.
Увеличение диаметра черепа происходит, главным образом, за счет затылочной области, в которой образуется киста, вызывающая истончение и растяжение костной основы. При выраженной гидроцефалии голова малыша активно растет на протяжении первых двух месяцев, параллельно происходит расхождение швов между костями спереди или в заднем отделе. Кроме того, характерны:
- Повышение нервной возбудимости (рефлексов);
- Глазодвигательные расстройства — нистагм, косоглазие;
- Приступы остановки дыхания;
- Парез лицевого нерва.
Симптомы мозжечковых нарушений у новорожденных выявить невозможно, и даже тяжелый дефект формирования мозжечковых структур далеко не всегда вызывает значимые признаки атаксии (двигательных расстройств), которая регистрируется всего у трети пациентов.
Значительно чаще, нежели двигательные нарушения, возникают расстройства психической деятельности, интеллекта, которые проявляются на фоне общей двигательной «неловкости». У 25% больных с гипоплазированным мозжечком имеются признаки аутизма, в связи с чем специалисты пытаются найти взаимосвязь между изменениями мозжечка и аутизмом у детей.
Дети с гидроцефалией в раннем возрасте беспокойны, плохо спят, характерен монотонный крик, усиление рефлексов, плавающие движения глаз и их закатывание, выраженность сосудов роговицы, заметная подкожная венозная сеть по мере роста размеров головки. Спонтанная двигательная активность новорожденных может быть ослаблена, возможны судороги и тетрапарез из-за гипертонуса мышц.
В более старшем возрасте становится заметным отставание в психическом и интеллектуальном развитии, дети не могут обучаться, быстро устают, плохо усваивают новую информацию, что делает процесс адаптации крайне затруднительным. В тяжелых случаях обучение невозможно совсем, в связи с чем ребенок нуждается в постоянной посторонней помощи, уходе и рассмотрении вопроса об инвалидности.
Моторное развитие заметно замедлено. При тяжелых формах аномалии дети не могут своевременно научиться переворачиваться, ползать, садиться и ходить, не удерживают взгляд на игрушках, быстро устают и часто плачут. Возможны нарушения питания с гипотрофией, общее снижение иммунитета, частые инфекционные заболевания.
Сочетание порока нервной системы с другими аномалиями развития органов предрасполагает к серьезным осложнениям, в числе которых не только мозговая дисфункция, слабоумие, судорожный синдром, но и сердечная недостаточность, склонность к пневмониям при пороках сердца, хроническая почечная недостаточность и уремия при врожденном поликистозе, что усугубляет явления отека мозга и может послужить причиной гибели пациента.
При тяжелой окклюзионной гидроцефалии смерть может наступить в раннем младенчестве от отека головного мозга, фатальных аритмий, остановки дыхания на фоне компрессии стволовых структур, тяжелой пневмонии и других инфекционных осложнений.
У взрослых возможно постепенное нарастание гидроцефалии с краниалгиями, снижением памяти и внимания, раздражительностью, склонностью к депрессии, утренней тошнотой и рвотой на высоте головной боли. В тяжелых случаях бывает судорожный синдром. Возможны проблемы с координацией и выполнением мелких движений, неуверенность при ходьбе, зрительные расстройства.
Диагностика и лечение
Диагностика синдрома Денди-Уокера основывается на результатах ультразвукового осмотра, при этом важно обнаружить аномалию еще во время эмбрионального развития. УЗИ становится информативным после 18 недели гестации, но в некоторых случаях заподозрить патологию можно и раньше — уже на 14-15 неделях эмбрионального развития.
Диагностическими критериями при аномалии задней черепной ямки считаются:
- Наличие крупной кистозной полости, включающей четвертый мозговой желудочек, в задней части черепа;
- Отсутствие или аномальное развитие червя мозжечка;
- Гипоплазия мозжечковых гемисфер, наличие широкой щели между ними;
- Расширение желудочковой системы (гидроцефалия).
Для постановки диагноза синдрома Денди-Уокера необходимы:
- УЗИ (нейросонография);
- МРТ для определения анатомических особенностей четвертого желудочка мозга;
- Консультация офтальмолога;
- Осмотр нейрохирурга;
- УЗИ сердца для исключения врожденных аномалий;
- Консультация генетика и определение кариотипа при возможных генетических мутациях.
Лечение патологии определяется симптоматикой и тяжестью проявлений. Если гидроцефалии нет, а внутричерепное давление в пределах нормы, то оправдано динамическое наблюдение невролога, педиатра или нейрохирурга, каких-либо медикаментов не требуется.
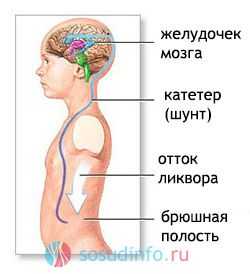
шунтирование для нивелирования гидроцефалии
При нарастании гидроцефалии и внутричерепного давления показаны шунтирующие хирургические операции для отвода ликвора из черепа в грудную или брюшную полость. Медикаментозное лечение включает применение диуретиков (диакарб, маннитол), ноотропных средств (пирацетам, пантогам), антиконвульсантов (депакин).
В случае гипертонуса показаны физиотерапевтические и водные процедуры, массаж, специальные упражнения. Важен тщательный уход и постоянное наблюдение за малышом, создание спокойной обстановки при беспокойном поведении и нарушениях сна.
При тяжелых формах течения патологии с отставанием в интеллектуальном развитии детям показана работа с дефектологами-педагогами, психологом по индивидуальной программе, исключающей избыток информации и умственное перенапряжение.
Прогноз при синдроме Денди-Уокера зависит от ряда причин: времени установления диагноза, наличия других пороков и хромосомных болезней, степени окклюзии ликворных путей. Смертность и заболеваемость после рождения выше в тех случаях, когда аномалия сочетается с другими дефектами и обнаружена до рождения малыша.
Гидроцефалия и внутричерепная гипертензия — ключевые моменты в определении прогноза, которые влияют и на развитие пациента, и на продолжительность и качество его жизни. В случае изолированного поражения мозга без признаков гидроцефалии прогноз благоприятный. Ребенок может развиваться по возрасту, а иногда аномалия и вовсе выявляется у взрослых при обследовании по поводу других причин.
В связи с тем, что причины порока так и не выяснены, проводить специфическую профилактику не представляется возможным. Конечно, нужно соблюдать здоровый образ жизни, особенно, женщинам, планирующим беременность или уже забеременевшим с исключением вредных привычек, неблагоприятных влияний внешней среды. Важно своевременно выявить и пролечить цитомегаловирусную инфекцию, герпес, а в случае краснухи, которой женщина заболела при беременности, врачи предложат аборт по медицинским показаниям из-за высокого риска сочетанных пороков.
Решать вопрос о сохранении беременности в том случае, если синдром возник случайно, у плода абсолютно здоровой женщины, придется будущей маме и ее семье. Решение всегда дается сложно, но следует знать, что аномалия нервной системы и нормальное развитие и рост ребенка — скорее, исключение из правил.
В абсолютном большинстве случаев детям и родителям приходится бороться с гидроцефалией, зачастую требуется не одна дорогостоящая и сложная операция, тогда как ее эффективность и прогноз все равно могут оставаться сомнительными.
Видео: примеры детей с синдромом Денди-Уокера
Синдром Жубера: признаки, фото МРТ, лечение, клинический случай
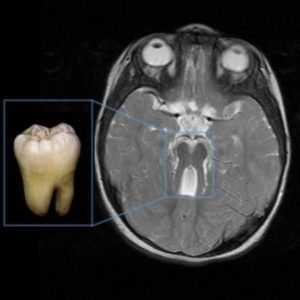
Редким, но сложным наследственным заболеванием является «Синдром Жубер». Затрагивает оно непосредственно нервную систему, вследствие недоразвития червя мозжечка, а так же ствол головного мозга. Эти центры нервной системы отвечают за координацию движений, равновесие, регулируют жизненно важные процессы в организме.
Основными признаками синдрома Жубера (Joubert syndrome) являются:
- нарушение в образовании и развитии мозжечка и ствола головного мозга, четко выявляемое на МРТ («синдром молярного зуба» или «molar tooth sign»);
- пониженный мышечный тонус (гипотония);
- задержка умственного и физического развития.
Так же могут наблюдаться нарушения во время дыхания (приступы апноэ и тахипноэ), аномалии движения глаз.
Этиология и наследование
Установлено, что возникновение болезни в организме человека связано с мутацией в 18 генах, а именно:
- NPHP1;
- AHI1 (у 12%);
- NPHP6;
- MKS3;
- RPGRIP1L;
- CC2D2A (у 9-10%);
- ARL13B;
- INPP5E;
- OFD1;
- TMEM216 (у 5%);
- KIF7;
- TCTN1;
- TCTN2;
- TMEM237 (у 2-3%);
- CEP290 (у 10%);
- TMEM138 (у 1-2% случаев);
- C5orf42.
Согласно научной статьи о синдроме Жубер, наблюдается около 50% случаев, когда генетические изменения происходят лишь в одном из вышеперечисленных генов.
По современной статистике данное генетическое отклонение встречается у одного из миллиона новорожденных. При этом абсолютно не имеют значение пол ребенка, его расовые особенности.
Нарушения в работе дыхательной системы связаны с недоразвитием стволовой части головного мозга. По этой же причине можно наблюдать беспорядочное движение языка и глаз, проявляется задержка в психомоторном развитии.
Название заболевания возникло от фамилии канадского педиатра Мари Жуберт, которая в 1969 году впервые описала его симптоматику. У ее четырех пациентов, родители которых были кровными родственниками, наблюдались нарушения слуха, ритмичности дыхательного процесса, слабость мышечной системы и интеллектуальное торможение. Сам термин «синдром Жубер» начал употребляться в медицине с 1977 года.
Причины мутации и факторы риска
В медицинской практике до конца не изучены все причины генной мутации, которые способствуют данному заболеванию. Однако точно известно, что патология передается от родителей по аутосомно-рецессивному типу в том случае, если оба родителя имеют видоизмененный ген.
Клиника и симптомы
Существует три формы протекания данного нарушения:
Со временем болезнь имеет прогрессирующий характер и сопровождает человека пожизненно.
Основные признаки синдрома Жубер проявляются с самого рождения:
- отклонения в развитии мозжечка и ствола головного мозга, которые можно наблюдать при проведении обследования МРТ;
- снижение кровяного давления и, как следствие, пониженный тонус мускулатуры;
- задержка в умственном развитии.
Лишние пальцы могут быть при некоторых вариантах генных мутаций
Синдром молярного зуба и другие признаки болезни Жубера на МРТ:
Среди дополнительных признаков выделяют:
- нарушения дыхательных процессов (апноэ, тахипноэ);
- глазодвигательная апраксия (беспорядочные движения глаз);
- нарушения в координации движений;
- частые судороги;
- патологические изменения сетчатки;
- наличие «заячьей губы»;
- полидактилия (наличие лишних пальцев на верхних и нижних конечностях);
- патологии в работе эндокринной, выделительной и других системах органов.
Клинический случай
В медицинских справочниках можно найти описание такого клинического случая:
В первые месяцы жизни наблюдалась гипотония мышц, не координированные движения глаз, нарушения ритма дыхания. В полтора года, после проведения ряда обследований, поставлен диагноз «Синдром Жубер». Во время плача у ребенка случались кратковременные эпизоды апноэ, закатывались глаза. Такие приступы были расценены как эпилептические, и далее ребенка лечили от эпилепсии. К 4 годам противоэпилептическая терапия была отменена в связи с проявившимися побочными эффектами.
Во время проведения МРТ в пятилетнем возрасте наблюдаются киста задней ямки черепа, а также «синдром коренного зуба». К 5 с половиной годам сохранились все признаки синдрома Жубер, а именно пониженный тонус мышц, неравномерное дыхание, отсутствие речевых и жевательных навыков, не координируемое движение глаз.
Диагностика заболевания
Помочь диагностировать нарушения в работе центральной нервной системы могут такие клинические исследования:
- полный визуальный осмотр;
- проведение генетического тестирования;
- МРТ (магнитно-резонансная томография);
- обследование внутренних систем органов, которые повреждены вследствие данной генной патологии.
Осмотр больного помогает определить мышечный тонус, нарушения в координации движений, снижение сухожильных рефлексов. Легко определяется отсталость в умственном развитии.
Диагностика с помощью МРТ продемонстрирует развитие отделов головного мозга. У пациентов с данным диагнозом наблюдаются изменения, напоминающие разрез зуба («синдром коренного зуба»). Это свидетельствует о патологических изменениях стволовой части мозга и червя мозжечка. Другими признаками недоразвития мозга являются расширенные желудочки, гидроцефалия, недоразвитие мозжечка.
При обследовании внутренних органов часто наблюдаются отклонения в работе сердца, лёгких, поликистозное заболевание почек, фиброз печени. Офтальмолог отмечает видоизменения в развитии сетчатки глаза, не координируемые движения органов зрения.
Проведение генетических тестов поможет определить наличие мутации в генах, которые передались по наследству.
Возможности современной медицины
Способов лечения на сегодняшний день не существует. Врачи могут лишь облегчить симптоматику заболевания. Так для уменьшения проявления неврологических отклонений применяют нейрометаболические стимуляторы, которые могут помочь в интеллектуальном развитии.
Методы физиотерапии, различные физические нагрузки помогут восстановить мышечный тонус, координацию движений.
Важно следить за дыханием больного, стимулировать дыхательную деятельность, чтобы исключить возможность апноэ.
Для формирования и постановки речи проводится работа со специалистами (логопед, дефектолог).
Прогноз и профилактика
Установить точный прогноз синдрома Жубера невозможно, так как все зависит от формы выраженной симптоматики и тяжести протекания болезни. В самых тяжелых случаях возможен летальный исход еще в младенчестве по причине апноэ или неврологических отклонений.
Чаще всего больной может дожить до глубокой старости, но при этом иметь инвалидность из-за нарушений зрения, слуха и других патологий внутренних органов. Умственное развитие возможно восстановить, но с явным торможением в интеллектуальных способностях.
Профилактическими действиями являются:
- перинатальная диагностика плода;
- выявление носителя патологической генной формы с тяжелой наследственностью;
- консультации специалистов при планировании беременности.
Иногда во время болезни возникают приступы, которые легко можно спутать с эпилептическим заболеванием. Однако ни в одном из описанных клинических случаев подтверждения эпилепсии не встречается.
Аномалия Арнольда-Киари: причины, синдром и симптомы, диагноз, лечение
Аномалию Арнольда-Киари относят к порокам кранио-вертебральной зоны. Формируется она в задней черепной ямке (ЗЧЯ), при недостаточном объеме которой происходит смещение задних отделов мозга и мозжечка в сторону большого затылочного отверстия, а также нарушается ток спинномозговой жидкости.
Задняя часть черепа образует так называемую заднюю черепную ямку, в которой расположены полушария и червь мозжечка, мост, продолговатый мозг, переходящий в спинной после прохождения сквозь большое затылочное отверстие. Большое затылочное отверстие ограничено костной основой черепа и не способно менять диаметр, любые смещения структур мозга чреваты несоответствием их размеров и диаметра отверстия, вклиниванием и ущемлением нервной ткани, последствия которого могут стать фатальными.
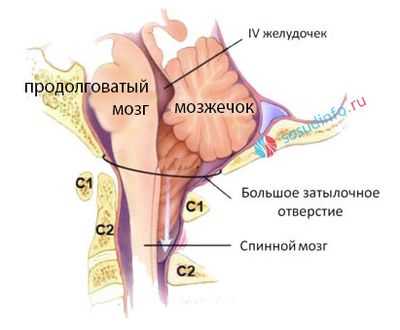
В продолговатом мозге сконцентрированы жизненно важные нервные центры, отвечающие за деятельность сердечно-сосудистой системы и дыхание, поэтому не только неврологический дефицит будет проявлением заболевания. В тяжелых случаях происходит угнетение витальных функций, и больной может умереть. Смещение гемисфер мозжечка влечет остановку циркуляции ликвора с гидроцефалией, которая еще больше усугубляет имеющиеся расстройства.
Аномалия Арнольда-Киари бывает врожденной, формирующейся у плода и сочетающейся с другими отклонениями в развитии, а клиника ее не всегда появляется сразу. В части случаев до манифестации патологии проходит значительный промежуток времени либо возникает ситуация, провоцирующая проявление бессимптомной до этого аномалии, у других пациентов и вовсе она может оказаться случайной находкой на МРТ. Нередко патология носит приобретенный характер и возникает под действием внешних причин, при этом мозг и череп при рождении имеют нормальное строение.
Причины и механизм развития порока задней черепной ямки (ЗЧЯ)
Единого мнения по поводу этиологии аномалии Киари нет. Ученые выдвигают различные теории, каждая из которых вполне обоснована и имеет право на существование.
Ранее аномалию считали исключительно врожденным пороком, однако наблюдения специалистов показали, что только малая часть пациентов имели дефекты во время внутриутробного развития, остальные же их приобрели уже в процессе жизни.
Причинами приобретенной патологии кранио-вертебрального перехода считают неравномерность скорости роста нервной ткани мозга и костной основы черепа, когда мозг увеличивается значительно быстрее, нежели костное вместилище, в котором он находится. Возникающее в конечном счете несоответствие объемов и служит основой болезни Арнольда-Киари.
Врожденная форма патологии сочетается с костной дисплазией, влекущей недоразвитие костей черепа, а также с нарушением формирования связочного аппарата, и любое внешнее воздействие, травма способны резко усугубить проявления патологии. Характерным считается сочетание порока черепной ямки с другими нарушениями утробного развития и врожденными синдромами.
Неврологами сформулировано два основных механизма формирования патологии:
- Уменьшение размеров ЗЧЯ при нормальных объемах отделов мозга (вероятно, вследствие нарушений во время внутриутробного периода).
- Увеличение объемов самого головного мозга при сохранении правильных параметров черепной ямки и большого затылочного отверстия, когда мозг оттесняет свои каудальные отделы в направлении затылочного отверстия.

Так как аномалия может быть врожденной, то среди причин указывают те, которые способны изменить нормальное течение беременности и внутриутробного развития:
- Злоупотребление медикаментами, прием алкоголя и курение при вынашивании плода, особенно — на ранних сроках, когда только формируются органы и системы зародыша;
- Вирусные поражения у беременных, среди которых особую опасность представляют инфекции с тератогенным эффектом — краснуха, цитомегаловирус и др.
Аномалия Арнольда-Киари возникает и по ряду приобретенных причин при изначально правильно развитых мозге и костях черепа. Привести к ее появлению уже после рождения способны:
- Родовые травмы, как спонтанные, так и при акушерских пособиях;
- Черепно-мозговые травмы и гидродинамический удар ликвора о стенки канала спинного мозга в случае нарушения ликвородинамики (так происходит у взрослых);
- Гидроцефалия.
Гидроцефалия может быть провоцирующим фактором, так как увеличение объема содержимого в черепе, пусть даже за счет жидкости, неминуемо влечет увеличение давления и смещение мозговых отделов в каудальном (заднем) направлении. С другой стороны, она является проявлением самой аномалии, когда опущение мозжечка вызывает блокаду ликворных путей и нарастание давления спинномозговой жидкости, циркулирующей по полостям мозга.
Типы и степени аномалии Арнольда-Киари
В зависимости от наличия тех или иных изменений со стороны мозга и костной основы черепа, принято выделять несколько разновидностей аномалии Арнольда-Киари:
- Аномалия Киари 1 типа, когда возникает перемещение книзу миндалин мозжечка, обычно выявляется у взрослых и подростков, нередко комбинируется с нарушением ликвородинамики и накоплением ликвора в центральном канале спинного мозга (гидромиелия). Возможно сдавливание мозгового ствола.
Аномалия Арнольда-Киари 1 типа – наиболее часто диагностируемая и имеющая довольно благоприятный прогноз
- Аномалия Арнольда-Киари 2 типа — манифестирует уже у новорожденных, так как происходит смещение значительно большего объема мозга, чем при 1 типе: миндалины мозжечка и его червь, продолговатый мозг с четвертым желудочком, возможно — образования среднего мозга. Обычно при 2 степени порока происходит нарушение тока ликвора с гидромиелией. Заболевание часто сочетается с наличием врожденной грыжи спинного мозга и аномалиями позвонков.
- 3 тип заболевания характеризуется выпячиванием мягкой мозговой оболочки с веществом мозга в затылочной области, куда попадают также мозжечок и продолговатый мозг.
аномалия Арнольда-Киари 3 типа на снимке
- Аномалия Арнольда-Киари 4 типа проявляется недоразвитием мозжечка, когда последний уменьшен, поэтому не опускается дистальнее к каналу в кости. Патология делает новорожденного нежизнеспособным и обычно заканчивается смертью.
Что касается степней тяжести, то:
Аномалию Арнольда-Киари 1 степени можно считать одним из наиболее легких вариантов патологии, так как пороков самого мозга при ней практически не возникает, а клиника может и вовсе отсутствовать, появляясь лишь при неблагоприятных условиях — травма, нейроинфекция и т. д.
Мальформации второй и третьей степени, в свою очередь, часто сочетаются с разнообразными пороками развития нервной ткани — гипоплазией некоторых участков мозга и подкорковых узлов, смещением серого вещества, кистами ликворных путей, недоразвитием извилин мозга.
Проявления синдрома Арнольда-Киари
Симптоматика синдрома Арнольда-Киари определяется его типом и характером смещения структур ЗЧЯ. Часто он протекает бессимптомно и обнаруживается случайно в ходе обследований мозга. У взрослых появление симптоматики может спровоцировать травма головы, у малышей некоторые формы заболевания заметны уже в первые часы и дни жизни.
Аномалия I типа диагностируется наиболее часто и может проявиться в подростковом либо взрослом возрасте следующими синдромами:
- Гипертензионным;
- Церебеллярным;
- Бульбарным;
- Сирингомиелическим;
- Явлениями поражения черепных нервов.
Гипертензионный синдром вызван увеличением внутричерепного давления вследствие блокады оттока ликвора сместившимися отделами мозга. Он проявляется:
- Головными болями в области затылка, особенно, при чихании, кашлевых толчках;
- Тошнотой и рвотой, после которой больной не ощущает облегчения;
- Напряжением мышц шеи.
Признаками вовлечения мозжечка (церебеллярный синдром) считают расстройства речи, двигательной функции, равновесия, нистагм. Пациенты жалуются на шаткость походки, неустойчивость положения тела в пространстве, затруднение мелкой моторики и четкости движений.
Поражение стволового отдела мозга представляется опасным ввиду расположения там ядер черепных нервов и жизненно важных нервных центров. Стволовая симптоматика состоит в:
- головокружении; двоении в глазах и снижении зрения;
- затруднении глотания;
- ухудшении слуха, шуме в ушах;
- обмороках, гипотонии, ночных апноэ.
Взрослые носители мальформации Арнольда-Киари указывают на нарастание головокружения и шума в ушах, а также пароксизмы потери сознания при поворачивании и наклонах головы. Вследствие сдавления стволов черепных нервов появляется атрофия половины языка и нарушение движения гортани с расстройством акта глотания, дыхания и голосообразования.
При образовании полостей и ликворных спинномозговых кист на фоне затрудненного тока спинномозговой жидкости у пациентов с I вариантом мальформации возникают признаки сирингомиелического синдрома — расстройство чувствительной сферы, онемение кожи, гипотрофия мышц, дисфункция тазовых органов, снижение и исчезновение брюшных рефлексов, периферические нейропатии и изменения со стороны суставов.
Расстройства чувствительности сопровождаются нарушением восприятия собственного тела, когда пациент, закрыв глаза, не может сказать, в каком положении находятся его руки или ноги. Снижается также чувствительность к боли и температуре.
По наблюдениям неврологов, диаметр и локализация кисты спинного мозга не обязательно отражаются на выраженности и распространенности нарушений чувствительной и двигательной сферы, гипотрофии мышц.
При синдроме второго и третьего типа течение патологии значительно тяжелее, симптоматика появляется у ребенка сразу же после родов. Характерны нарушения дыхания — стридор (шумное дыхание), приступы его остановки, а также двусторонний парез гортани, провоцирующий расстройства глотания, когда жидкая еда попадает в носовые ходы.
Второй тип аномалии у малышей первых месяцев жизни сопровождается нистагмом, усилением тонуса мышц в руках, синюшностью кожи, которые особенно заметны при кормлении грудничка. Двигательные нарушения вариабельны, проявления их меняются, возможна тетраплегия — паралич и верхних, и нижних конечностей.
Аномалия Арнольда-Киари третьего и четвертого вариантов протекает тяжело, это врожденная патология, которая не совместима с нормальной жизнедеятельностью, поэтому прогноз при таком диагнозе нельзя считать благоприятным.
Мальформация Арнольда-Киари может приводить к осложнениям, вызванным блокадой тока ликвора, поражением ядер черепных нервов, ущемлением стволовых структур. Самыми частыми считаются:
- Гипертензионно-гидроцефальный синдром — увеличение внутричерепного давления вследствие блокады путей ликворооттока, возможен как у детей, так и у взрослых;
- Расстройства дыхания, апноэ;
- Инфекционно-воспалительные процессы — бронхопневмонии, уроинфекции, которые связаны с лежачим положением пациента, нарушением актов глотания и дыхания, функции тазовых органов.
При тяжелом течении патологии может наступить кома, остановка сердца и дыхания, которые приводят к смерти в считанные минуты. Реанимационные мероприятия позволяют обеспечить витальные функции, но вернуть к жизни мозг и устранить необратимые последствия компрессии его отделов, к сожалению, практически невозможно.
Диагностика и лечение мальформации Арнольда-Киари
По особенностям симптоматики и на основании осмотра невролога диагноз мальформации Киари поставить невозможно. Энцефалография, исследования сосудов головы тоже не дадут никакой информации касательно причин неврологических нарушений, однако могут показать наличие повышенного давления в черепе. Рентгенография, КТ, МСКТ укажут на наличие дефектов костей черепа, которые характерны для этой патологии, но состояние мягкотканных структур, нервной ткани установить при этом нельзя.
Точная диагностика аномалии стала возможна благодаря использованию МРТ, посредством которой врач может определить и костные пороки, и варианты развития самого мозга, его сосудов, уровень расположения отделов относительно черепных костей, их размеры, объем задней черепной ямки и ширину большого затылочного отверстия. МРТ можно считать единственным точным и самым достоверным методом выявления патологии.
МРТ требует обездвиживания пациента, который должен какое-то время спокойно лежать на столе аппарата. У детей с этим могут возникнуть значительные трудности, поэтому исследование проводят в состоянии медикаментозного сна. Для поиска сочетанных пороков спинного мозга и позвоночника исследуют также эти отделы позвоночного столба.
Когда диагноз установлен, пациента направляют к нейрохирургу или неврологу для определения плана лечения, показаний к хирургической операции, ее вида.
Аномалия Арнольда-Киари, протекающая бессимптомно, не требует лечения. Более того, и сам носитель патологии может не догадываться о том, что в организме что-то не так. При появлении клинических признаков заболевания показано консервативное или оперативное лечение.
Если проявления ограничены головными болями, назначается медикаментозная терапия, включающая противовоспалительные средства (найз, ибупрофен, диклофенак), анальгетики (кеторол) и препараты, снимающие мышечный спазм (мидокалм).
При наличии неврологических расстройств, признаков сдавления отделов мозга, нервных стволов, в случае отсутствия эффекта от медикаментозного лечения в на протяжении 3 месяцев, больному необходима хирургическая коррекция.
Операция необходима для устранения сдавливания нервной ткани и нормализации циркуляции ликвора. Самой популярной операцией при заболевании Киари считается краниовертебральная декомпрессия, которая направлена на увеличение размеров ЗЧЯ.
При декомпрессии хирург удаляет участки затылочной кости, резецирует миндалины мозжечка, по необходимости производит иссечение задних отделов первых шейных позвонков. Для предупреждения пролабирования задних частей мозга в полученное отверстие на твердую мозговую оболочку накладываются специальные синтетические заплаты.
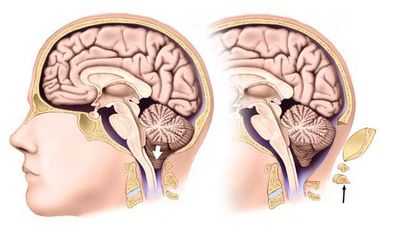
пример удаления частей затылочкой кости и шейных позвонков
Декомпрессия ЗЧЯ считается и травматичной, и рискованной. Статистика такова, что осложнения возникают как минимум у каждого десятого больного, в то время как без операции показатель смертности намного ниже. В связи с высоким риском оперативного лечения нейрохирурги прибегают к нему только в случае действительно серьезных показаний — клиники сдавления участков мозга.
Другим вариантом хирургического лечения считается шунтирование, которое обеспечивает отток ликвора из полости черепа в грудную или брюшную полость. Путем имплантации специальных трубок происходит переток спинномозговой жидкости и снижение внутричерепного давления.
При тяжелых формах патологии показана госпитализация, мероприятия по профилактике инфекционных осложнений и коррекции неврологических расстройств. Нарастание отека мозга вследствие вклинивания его отделов в затылочное отверстие требуют лечения в условиях реанимации, включающего борьбу с отеком (магнезия, фуросемид, диакарб), налаживание искусственной вентиляции легких при нарушении дыхания и т. д.
Продолжительность жизни и прогноз при аномалии Арнольда-Киари зависят от типа патологии. При I типе прогноз можно считать благоприятным, в ряде случаев клиника не возникает совсем либо провоцируется лишь сильными травмирующими факторами. При бессимптомном течении носители аномалии живут столько же, сколько и все остальные люди.
При аномалии второго и первого типа с клиническими проявлениями прогноз несколько хуже, так как проявляется неврологический дефицит, который сложно устранить даже при активном лечении, поэтому для таких пациентов большое значение имеет своевременно проведенная операция. Чем раньше больному будет оказана хирургическая помощь, тем менее выраженные неврологические изменения его ждут.
Мальформация третьего и четвертого типов — наиболее тяжелые формы патологии. Прогноз неблагоприятен, так как вовлекаются многие структуры мозга, часто имеют место сочетанные пороки других органов, тяжелые нарушения функции ствола мозга, не совместимые с жизнью.
Читайте также: