Микроцефалия (микрэнцефалия) - клиника, диагностика
Добавил пользователь Валентин П. Обновлено: 30.01.2026
Микроцефалия (микрэнцефалия) - клиника, диагностика
Микроцефалию иногда определяют как уменьшение окружности головы более, чем на два стандартных отклонения ниже среднего (Barkovich et al., 2001а) или менее 3-го центиля (Haslam, 2000), однако Barkovich et al. (2001а) определили ее как «окончательную микроэнцефалию». Оба определения, очевидно, включают нормальных индивидуумов. Микрэнцефалия, с другой стороны, теоретически относится к маленьким размерам мозга (Friede, 1989), но на практике оба термина равноценны.
Все авторы допускают, что дети с умеренно маленькими головами (между 2 и 3 СО ниже среднего) часто нормальные, но статистически отмечается преобладание легкой микроцефалии среди детей с трудностями в обучении. Задержка умственного развития наиболее характерна, когда небольшие размеры головы сопровождаются задержкой развития. Однако даже пациенты с окружностью головы между 3 и 4 СО ниже среднего могут иметь нормальный интеллект.
Этиология микроцефалии разнообразна (Opitz и Holt, 1990). Многие случаи, называемые вторичной микроцефалией, являются результатом приобретенных кластических повреждений или других патологических процессов во время внутриутробного развития или позднее, в первые годы жизни, во время быстрого роста головы и мозга (Hughes и Miskin, 1986). Существенная доля случаев, несомненно, имеет первичный характер, в основном генетического происхождения. Причины подобных случаев почти неизвестны.
Недостаточная клеточная пролиферация является лишь одним из этих механизмов, потенциально способным вызывать чрезмерную гибель эмбриофетальных клеток. Генетическая микроцефалия может принадлежать к нескольким видам. Sugimoto et al. (1993) обнаружили, что 44 из 55 случаев имели генетическое происхождение, остальные были связаны с различными негенетическими ошибками развития.
Первичная микроцефалия (также называемая «истинной» микроцефалией или «microcephalia vera») наследуется по рецессивному типу (Suri, 2003). Частота варьирует в зависимости от популяции с определенным влиянием распространенности близкородственных браков. Степень микроцефалии обычно значительная (3 СО и часто 5-6 СО ниже среднего), узкий выступающий лоб и заостренное темя демонстрируют своеобразную клиническую и рентгенологическую картину. Парадоксально, при microcephalia vera отсутствуют тяжелые неврологические симптомы, но нередки гиперкинетическое поведение и расстройства тонкой двигательной координации (Accardo и Whitman, 1988).
В типичных случаях судороги не развиваются. Имеет место некоторая степень задержки умственного развития, но она обычно легкая и большинство детей могут освоить, по крайней мере, упрощенный язык. Все это становится более заметным, когда во взрослом состоянии вес мозга становится менее 500 г.
С патологической точки зрения, извилины мозга выглядят нормальными и кора истончена. Отмечается тяжелое истощение нейронов в II и III слоях (Evrard et al, 1989).

Микроцефалия
Mirocephalia vera генетически гетерогенная. Как правило, имеет место рецессивный тип наследования. Были картированы шесть локусов (МСРН1-5) и определены два гена (Suri, 2003). В трети семей, обследованных Suri, не было продемонстрировано связей, определяющих дальнейшую гетерогенность. Третий ген недавно был выявлен при похожем состоянии распространенном в секте амишей, американских меннони-тов. Ген в МСРН1 локусе кодирует белок, микроцефалии, связанный с ростом мозга. У гетерозигот ген может вызывать легкую умственную отсталость, иногда с относительно маленьким размером головы.
В других случаях микроцефалии кора мозга патологически истончена с небольшим количеством аномальных извилин. Эти случаи были названы микролиссэнцефалией (Barkovich et al., 1998) или олигирической микроцефалией (Dobyns и Barkovich, 1999). Вероятно существует несколько подтипов (Ross et al., 2001). Они клинически отличаются от первичной микроцефалии наличием глубокой задержки умственного развития, судорогами и неврологической симптоматикой, а иногда зрительной и другой патологией (Silengo et al., 1992, Kroon et al., 1996).
MPT мозга показывает различные аномалии извилин коры помимо небольших размеров мозга и плохо развитых лобных долей (Steinlin et al., 1991, Sugimoto et al., 1993, Sztriha et al., 2004). К другим редким типам мироцефалии относится мозжечковая гипоплазия (Kato et al., 1999, Ross et al„ 2001, Barth, 2003a) и синдром Неу-Лаксова (Manning et al., 2004).
Эти случаи являются частью гетерогенной группы «микроцефалии плюс». Tolmie et al. (1987) обнаружили, что только в 1 из 29 исследованных случаев имелась «microcephalia vera», тогда как в остальных клинические проявления были сложнее, включая судороги и/или спастичность. Большинство случаев были, несомненно, рецессивными (Harbord et al., 1989). Другие типы могли иметь доминантный характер наследования (Burton, 1981). Однако встречалось и множество ассоциированных аномалий, включая врожденный нефротический синдром (Nishikawa et al., 1997), короткую тощую кишку (Nezelof et al, 1976), агенезию мозолистого тела и тяжелые расстройства миграции (King et al., 1995).
Семейные случаи с внутричерепной кальцификацией, маскирующейся под внутриутробную инфекцию (синдром Айкарди-Гутиере), представляет особый интерес из-за сходства с пренатальными инфекциями, поэтому диагностика семейного состояния в этих случаях обычно упускается (Baraitser et al., 1983).
Другой конец спектра представлен редкими случаями клинически незаметной, передающейся по доминантному типу, микроцефалии (Haslam и Smith, 1979, Accardo и Whitman, 1988).
Случаи с наличием или без расстройств неврологического развития с хромосомной нестабильностью (Seemanova et al., 1985) или первичной комбинированной иммунной недостаточностью (Berthet et al., 1994) встречаются редко. Исчерпывающий перечень синдромов с микроцефалией представил Friede (1989).
Крайний вариант микроцефалии — радиальный микромозг. По Evrard et al. (1989), вес мозга в таких случаях может быть менее 16-50 г. Колонки радиального мозга почти нормальные, но их число значительно снижено. Такие случаи следует отличать от ателэнцефалии, при которой кортикальные структуры отсутствуют.
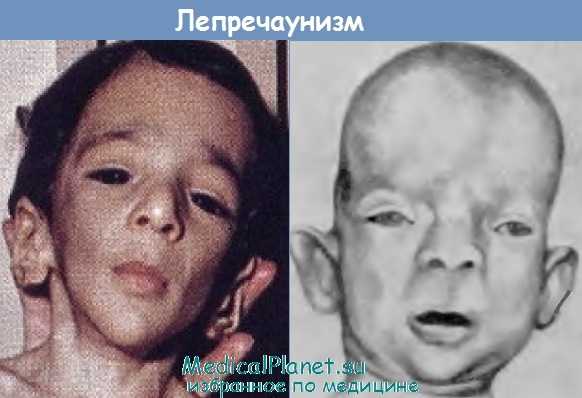
Микроцефалия при лепречаунизме
Микроцефалия также является признаком множества хромосомных аномалий, дисморфичного синдрома с умственной отсталостью и синдромов с карликовостью, такого как синдром Секкеля. Патология этих заболеваний обычно плохо изучена. Некоторые из этих синдромов имеют генетическое происхождение.
Диагноз микроцефалии прост при крайних проявлениях, но может быть сложен при легких формах. Размер головы в норме различается в семьях и индивидуально. Измерение размеров головы является неточным и обычно используемая затылочно-лобная окружность не вполне коррелирует с объемом мозга, поэтому целесообразно измерение при КТ. (Gooskens et al, 1989). Однако измерение окружности головы остается простым распространенным методом оценки размеров мозга. У детей окружность головы быстро нарастает по мере роста мозга, поэтому соответствующая кривая роста должна использоваться у новорожденных и детей раннего возраста по средней массе при рождении, а также у больных и здоровых недоношенных новорожденных (Gross et al., 1983).
У здоровых детей масса мозга утраивается в течение первого года жизни, поэтому при обследовании обязательны повторные измерения.
Дифференциальная диагностика с тотальным краниосиностозом не является проблемой. Форма черепа, экзофтальмия, нормальное развитие, признаки повышенного внутричерепного давления и рентгенография в стандартных проекциях черепа делают диагноз очевидным. Основной задачей диагноза является дифференцирование приобретенной (кластической) микроцефалии от генетических типов. Выраженный неврологический компонент, тяжелая умственная отсталость и анамнез патологических событий в пренатальном или перинатальном периодах при средней микроцефалии указывают на кластическое происхождение, а противоположные признаки относятся к генетической микроцефалии.
Однако клинические проявления в значительном объеме связаны с неопределенностью. Диагностическую ценность представляет нейровизуализация деструктивных изменений. Напротив, нормальные результаты КТ не исключают возможность приобретенного характера заболевания.
Антенатальная диагностика может быть затруднена, особенно при отсутствии сопутствующих аномалий развития. Простое измерение бипариетального диаметра недостаточно, поскольку он может быть низким у плодов с сагиттальным краниосиностозом или из-за значительного внутриутробного моделирования головки. Важны повторные измерения с особым вниманием к форме черепа.
Прогноз при микроцефалии варьирует. Некоторые дети имеют относительно хороший интеллектуальный потенциал, и длительные усилия, прилагаемые к их обучению, ненапрасны. Специальное обучение неизбежно, хотя в исключительных случаях преуспевают дети даже с окружностью головы менее 6 СО ниже среднего (личное наблюдение).
Классификация нарушений развития коры головного мозга
Патологическое развитие коры головного мозга является весьма распространенной причиной расстройств нейроразвития. Высокая частота таких аномалий стала очевидной с развитием современных методов нейровизуализации, особенно МРТ, а роль в качестве причины эпилепсии, церебрального паралича и задержки умственного развития в настоящее время оценивается выше перинатально приобретенных повреждений. Используемые для описания этих аномалий термины разнообразны и неточны. Определения патологии нередко применяются для обозначения диагностированных методами визуализации состояний.
Такой термин как «корковая дисплазия» применяется как в общем значении, относящемся к любому типу аномального развития коры мозга, так и в более ограниченном смысле при расстройствах корковой организации. Термин «расстройства миграции» часто неправильно относят к патологии, ограниченной окончательной организацией коры.
Во избежание разночтений в данной главе определение аномалии развития коры относится ко всем типам. Широко распространено понятие «мальформации кортикального развития» со сходным значением (Barkovich et al., 1996, 2001а), но мальформации являются результатом неверного развития и соотнесение с процессом развития представляется сложным. Термин «нарушения миграции» и «нарушения организации» применимы только для патологии соответствующих стадий кортикогенеза.
Разработано несколько систем классификации корковых аномалий. Наиболее часто применяют классификацию Barkovich et al. (1996, 2001а с подразделением мальформаций на три группы в соответствии с предполагаемым временем определения и механизмами формирования различных типов: расстройства неврологической пролиферации/дифференцировки и апоптоза; мальформации в связи с патологией миграции; и мальформации в связи с патологической организацией коры (включая позднюю нейрональную миграцию). В таблице ниже представлена упрощенная форма этой классификации. Другие системы предлагают основываться на патологии, клинических проявлениях или генетических и молекулярных процессах (Sarnat и Flores-Sarnat, 2001b, 2002,2004; Clark, 2004).
С клинической точки зрения, полезна простая схема, разделяющая диффузные и очаговые корковые мальформации. Диффузные аномалии (например, лиссэнцефалия) отвечают за генерализованные судороги, особенно инфантильные спазмы, тяжелые неврологические симптомы и задержку умственного развития; очаговые аномалии (например, очаговые корковые дисплазии) в большей степени отвечают за очаговую эпилепсию, обычно с ранним началом (Fauser et al., 2006), не обязательно с познавательным или неврологическим дефицитом и лучшим исходом. Для диффузной корковой патологии возможна только симптоматическая терапия, тогда как эффективное хирургическое лечение приемлемо во многих случаях очаговой дисплазии.
Некоторые диффузные аномалии могут развиваться в связи с метаболическими дефектами (например, синдром Цельвегера) (Raoul et al., 2003). Многоочаговые дисплазии могут также давать картину похожую на таковую при диффузных пороках.

Классификация Barkovich et al. схематически показывает наиболее вероятный механизм для каждого типа корковой мальформации. Различные стадии кортикогенеза, описанные выше, в действительности взаимосвязаны и накладываются один на другой. Превращение клеток-предшественников определяется до их миграции (McConnell и Kaznowski, 1991), и эта детерминированность имеет отношение к последующим процессам миграции и организации. Так, патологические клетки, например, при туберозном склерозе или при некоторых типах «корковой дисплазии», часто не перемещаются в направлении их нормального расположения и/или образуют недостаточно нормальных связей.
С патологической миграцией сходным образом связаны расстройства клеточной миграции, например, при пахигирии или агирии. В результате, часто трудно определить этап нарушения коркового развития в конкретном случае, и возможно произвольное расширение классификации. Так, гемимегалэнцефалия и микроцефалия зачастую связаны с патологической миграцией и субкортикальные гетеротопии часто сосуществуют с корковыми аномалиями. Отдельные аномалии часто одновременно присутствуют в одном мозге и микродисгенезии (Meencke и Veith, 1992) встречаются совместно с субкортикальной и кортикальной патологией развития. Некоторые расстройства трудно подогнать под предложенную схему; к примеру, большая доля случаев микроцефалии или макроцефалии может быть не результатом нарушения развития коры, а следствием деструкции или не относящихся к патологии механизмов. Более того, даже постулируемые при некоторых дефектах развития механизмы в основном являются теоретическими, а многие аномалии обусловлены связью и взаимодействием ряда известных и неизвестных механизмов. Последовательно рассмотрим различные аномалии и их механизмы.
а) Расстройства пролиферации/дифференцировки коры головного мозга. В данном разделе основное внимание сосредоточено на случаях с основной патологией в виде нарушений пролиферации и дифференцировки, хотя изолированное проявление встречается редко. Феномен запрограммированной гибели клеток гипотетически затрагивает 30-50% образующихся нейронов и может сыграть важную роль, в то время как деструктивные процессы могут вызывать утрату объема мозгового вещества и с трудом дифференцироваться от недостаточности развития из-за особенностей восстановительных процессов в развитии плода (Marin-Padilla, 2000).
Туберозный склероз, который принадлежит к категории заболеваний пролиферации/дифференцировки, описан в отдельной статье на сайте в связи с преобладанием кожных проявлений.
б) Расстройства миграции клеток коры головного мозга. Клиническая значимость этих мальформаций велика в связи с их ролью во многих случаях задержки умственного развития и эпилепсии. Более того, они часто излечимы, особенно при очаговой патологии, которая может реагировать на хирургическое вмешательство (Barth, 2003а). Эти расстройства чаще являются составной частью более сложных процессов не только с нарушением миграции клеток, но и с разнообразным сочетанием с аномалиями дифференцировки и организации коры.
В клеточную миграцию могут вмешиваться приобретенные или генетические причины (Barkovich et al., 1996, 2001а; Lambert de Rouvroit и Goffinet, 2001, Gressens, 2006). В зависимости от времени, типа и тяжести воздействия причинного фактора, спектр нарушений миграции может проявляться в виде большой формы (гетеротопии, лиссэнцефалии-пахигирии или полимикрогирии) или малой формы (патологическое расслоение коры, микродизгенезии). Обе формы могут быть локализованными, многоочаговыми или диффузными и в разной степени сочетаться (Barkovich et al., 1996, Dobyns и Leventer, 2003).
Локализованные области патологии миграции часто связаны с более диффузными изменениями в мозге (Sisodiya et al., 1995). Возрастает количество идентифицированных генетических нарушений миграции, что может быть вызвано хромосомными делециями или генными мутациями. Они могут оказывать влияние на многочисленные, еще плохо изученные механизмы, особенно сигнальные системы, отвечающие за передвижения клеток по глиальным волокнам, адгезивных молекул между нейронами и глиальными проводниками и определение сигналов, контролирующих связность и увеличение функции колбочковидных зрительных клеток и необходимую для движения энергию.
Для формирования надлежащей архитектоники коры важны множественные гены, из которых известны лишь немногие, но возможно влияние и других факторов, особенно нервной активности для синаптической стабилизации, приводящей к организации функционирующей невральной цепи.
Известные или предполагаемые приобретенные причины охватывают внутриутробные инфекции, особенно цитомегаловирусную болезнь и циркуляторные нарушения. Причиной могут стать любые патологические процессы, затрагивающие глиальные проводники (например, кровоизлияние, инфаркт, инфекция). Некоторые патологические процессы, ранее считавшиеся результатом отклонений развития кортикальной пластинки, в действительности вызваны деструктивными процессами, возникающими после завершения основных этапов кортикогенеза (Williams et al., 1984, Evrard et al., 1989, Marin-Padilla, 2000, Barth, 2003b).
Гетеротопии являются скоплениями нейронов, которые не достигли своих нормальных мест расположения. При субэпендимарных гетеротопиях нейроны остаются в местах, соответствующих примитивным зонам пролиферации или задерживаются на своем пути к кортикальной пластинке (рис. 2.15). С другой стороны, гетеротопии могут быть результатом отсутствия стоп-сигнала с последующим избыточным образованием клеток в перивентрикулярной зоне (Tassi et al., 2005).
Перивентрикулярные гетеротопии часто являются частью сложных синдромов мальформации, таких как синдром Айкарди (Aicardi, 1996, 2005) и синдром птеригий (Holtmann et al., 2001), или ассоциированы с задними цефалоцеле (Roelens et al., 1999). Для редко встречающейся большой формы также характерны множественные аномалии миграции с особенно сильным вовлечением базальных ядер, что приводит к частично неразделенному мозгу с невидимыми желудочками (Shaw и Alvord, 1996).
Изолированные гетеротопии могут оставаться бессимптомными или давать начало эпилепсии или неврологическим симптомам (Barkovich и Kjos, 1992). Они могут располагаться субкортикально или в субэпендимальной области. Субэпендимальные гетеротопии были обнаружены у 2% пациентов с эпилепсией, составляя 20% случаев пороков развития коры с эпилепсией (Raymond et al, 1994). Они располагались преимущественно в задней части мозга и могли быть одно- или двусторонними (Dubeau et al., 1995, Aghakhani et al, 2005, Tassi et al., 2005). При одностороннем процессе они обычно спорадические и клинически проявляются в виде очаговых эпилептических приступов. Эпилептическая активность может возникать из самих узелков или из близлежащей коры.
Резекция области, определенной инвазивной ЭЭГ, независимо от того, расположена она в очагах или в перифокальной коре (Tassi et al. 2005), обеспечивает контроль за судорогами.
в) Расстройства кортикальной организации. В соответствии с классификацией Barkovich, эта категория включает некоторые нарушения поздней миграции. Как и в других категориях, некоторые из включенных состояний зависят от механизмов, отличных от нарушения организации. Во многих случаях очаговой кортикальной дисплазии, например, определяются патологические клетки как неврального, так и глиального происхождения, указывая на аномалию клеточной дифференцировки.
г) Аномалии, часто связанные с расстройствами развития коры. Некоторые пороки развития часто сопутствуют расстройствам миграции. Однако они также могут возникать изолированно или в сочетании с другими мальформациями. Среди них выделяют агенезию мозолистого тела, шизэнцефалию («истинную» порэнцефалию), септо-оптическую дисплазию и кольпоцефалию.
Макроцефалия, мегалэнцефалия - клиника, диагностика. Отличия макроцефалии от гидроцефалии
У 75% из них наблюдалась гидроцефалия и повышенное внутричерепное давление, у 3% — специфические синдромы и у 20% отмечалась первичная мегалэнцефалия с нормальным давлением. Только у 13% из последней группы развилась умственная отсталость или неврологические отклонения. Основные диагностические критерии — гидроцефалия или перицеребральные скопления, поскольку они делают необходимым немедленное лечение.
Соотношение мальчиков и девочек 4:1, а у 50% пациентов макроцефалия отмечается в семейном анамнезе.
Макроцефалия представлена двумя основными группами: мегалэнцефалия и гидроцефалия с перицеребральным скоплением. Последняя группа описана в отдельной статье на сайте.

Мегалэнцефалия. Под мегалэнцефалией подразумевается увеличение массы мозга. Некоторые авторы (Friede, 1989) отделяют пациентов с большими размерами головы, но нормальным процессом развития и функционирования нервной системы, от случаев «истинной» мегалэнцефалии, т.е. существенного увеличения мозга по сравнению с возрастной нормой. Только последняя группа принадлежит к расстройствам развития мозга. Это различие несколько преувеличено, так как большая голова — и даже тяжелый мозг при аутопсии — нередко встречается у нормальных лиц или в сочетании с заболеваниями, такими как нейрофиброматоз, без каких-либо признаков мозговой дисфункции.
Предметом описания в данной главе будет истинная мегалэнцефалия с чрезмерными размерами мозга, связанными с патологией развития, что вызывает как увеличение числа невральных элементов, так и невральных и глиальных, или нарушение миграции и организации (Friede 1989); оба типа аномалии часто сосуществуют. Во многих случаях также выявляют гигантские патологические клетки, напоминающие таковые при туберозном склерозе и свидетельствующие о нарушении клеточной пролиферации. Причиной могут быть и иные патологические процессы, к примеру, патологическое накопление или рост отсуствующих в норме не-невральных элементов, как при болезни Александера. Структура извилин в таком мозге часто изменена.
Даже при строгом определении мегалэнцефалия включает несколько отличающихся патологических и клинических форм. Она может быть частью специфических синдромов, таких как синдром Сотоса или многочисленных нейрокожных синдромов, таких как макроцефалия-мраморная кожа (Giuliano et al., 2004), или может быть изолированной. Механизм мегалэнцефалии в настоящее время плохо понятен. Нарушена регуляция клеточной пролиферации субопухолевого (гамартоматозного) происхождения, либо диффузного, либо более локализованного, что в некоторых случаях трудно дифференцировать от ганглиоглиом. Повышенный уровень инсулиноподобного фактора роста (ИФРII) был выявлен в мозге новорожденных с макроцефалией вместе с сильно нарушенным развитием неокортекса (Schoenle et al., 1986).
Клинические проявления крайне различаются, зачастую они могут отсутствовать совсем. У большинства пациентов отмечают задержку умственного развития и у многих — судороги и диффузные неврологические симптомы.
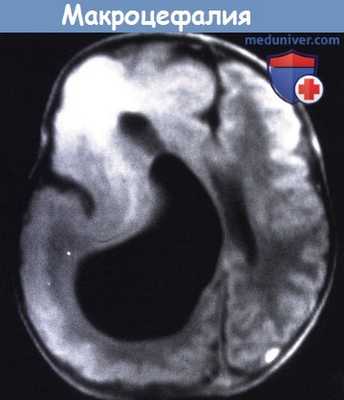
Т1 - взвешенное изображение МРТ пациента в возрасте пяти месяцев.
Большое правое полушарие, с патологически утолщенной корой и несколькими бороздами.
Значительно увеличенный желудочек и аномальный сигнал от лобного белого вещества.
Дифференциальный диагноз может быть трудным. Гидроцефалия и внутричерепные скопления должны быть исключены, поскольку требуют хирургического лечения. Дифференцировка мегалэнцефалии от больших размеров головы у детей с нормальным или практически нормальным развитием нервной системы обычно не вызывает затруднений. Однако такие дети составляют большую гетерогенную группу. У большинства из них, вероятно, большие размеры головы генетически детерминированы. Lorber и Priestley (1981) обнаружили высокую частоту макроцефалии у родителей, особенно отцов таких детей.
De Myer (1972) и Day и Schutt (1979) также предположили доминантный тип передачи «простой» макроцефалии. Некоторые из этих макроцефалических детей имели специфические нарушения способности к обучению, превышающие показатели детей с нормальным размером головы. Скорость увеличения размеров головы особенно высока у детей до четырех месяцев жизни (De Myer, 1972, Lorber и Priestley, 1981). На этой стадии часто возникают подозрения на гидроцефалию и, если темпы прироста быстро не снижаются, показана рентгенография. При отсутствии какой-либо патологии развития нервной системы и большого размера головы в семейном анамнезе не требуется ничего, кроме внимательного наблюдения. Однако необходимо исключить возможность пресимптоматической глутаровой ацидурии (Hoffmann et al., 1991) или другой органической ацидурии, часто сопровождающих макроцефалию.
У детей с долихоцефалией (синостоз сагиттального шва) важную роль играет форма черепа, потому что при выраженном удлинении черепа окружность головы у таких пациентов часто 3 СО и выше среднего, но сама по себе форма черепа вызывает подозрения. У маленьких детей с перинатальными заболеваниями или более старших, имеющих проблемы в питании, «догоняющий» рост временно осложняет диагностику (Sher и Brown, 1975). Нейровизуализацию необходимо выполнить в случаях выраженных расходящихся изгибов головы или при наличии симптомов повышенного внутричерепного давления.
Лечения мегалэнцефалии не существует, а в большинстве случаев в нем нет необходимости.
Микроцефалия
Микроцефалия – недоразвитие черепа и головного мозга, сопровождающее умственной отсталостью и неврологическими отклонениями. Микроцефалия характеризуется малыми размерами черепа, ранним смыканием черепных швов и закрытием родничка, судорожным синдромом, задержкой моторного развития, интеллектуальным дефектом, недоразвитием или отсутствием речи. Диагностика микроцефалии основывается на данных антропометрии, краниографии, КТ и МРТ головного мозга, ЭЭГ, НСГ; возможно пренатальное выявления микроцефалии у плода. При микроцефалии проводится симптоматическое лечение и реабилитационные мероприятия, направленные на социализацию ребенка.
МКБ-10
Общие сведения
Микроцефалия (микрокефалия) – тяжелый порок развития ЦНС, в основе которого лежит уменьшение массы головного мозга и уменьшение окружности черепа более чем на два-три сигмальных отклонения по сравнению со средними половозрастными показателями. Различные формы микроцефалии встречаются с частотой 1 случай на 10000 детей, в равных соотношениях среди мальчиков и девочек. Микроцефалия является причиной олигофрении в 10% наблюдений. При рождении окружность головы у ребенка с микроцефалией, как правило, не превышает 25-27 см (при норме - 35-37 см), а масса головного мозга – 250 г (в норме около 400 г).
Причины микроцефалии
С учетом времени и причин возникновения в педиатрии и детской неврологии выделяют первичную (наследственную, истинную) и вторичную (синдромальную и эмбриопатическую) микроцефалию. Первичная микроцефалия является компонентом наследственных болезней с аутосомно-рецессивным и рецессивным, сцепленным с полом типами наследования (синдром Джакомини, синдром Пейна). На долю истинной микроцефалии приходится 7-34% от всех форм патологии.
Вторичная микроцефалия отмечается при хромосомных аберрациях, наследственных энзимопатиях (фенилкетонурии), патологии беременности и родов. Синдромальная микроцефалия встречается более чем при 125 хромосомных аномалиях, наиболее частыми из которых являются болезнь Дауна (трисомия по 21 хромосоме), синдром Эдвардса (трисомия по 18 хромосоме), синдром Патау (трисомия по 13 хромосоме), синдром «кошачьего крика» (моносомия 5р) и др.
Вторичная эмбриопатическая микроцефалия обусловлена воздействием на плод тератогенных факторов и может являться следствием внутриутробных инфекций (краснухи, цитомегаловирусного энцефалита, герпеса, токсоплазмоза) и интоксикаций (алкогольной, наркотической, профессиональной), радиационного влияния, гипоксии, внутричерепных родовых травм, метаболических нарушений, гормональных заболеваний матери (сахарного диабета, тиреотоксикоза).
Микроцефалия у детей часто сочетается с другими аномалиями: расщелинами губы и неба («заячьей губой» и «волчьей пастью»), несовершенным остеогенезом, врожденной катарактой, пигментным ретинитом, первичной кардиомиопатией, лимфедемой, врожденными пороками сердца и легких, гипоплазией почек, что в значительной мере отягощает прогноз.
Патоморфологическое исследование головного мозга при микроцефалии выявляет уменьшение его массы свыше 25% от нормы, недоразвитие больших полушарий, особенно лобных отделов. При микроцефалии могут иметь место явления микро- или макрогирии (аномально узких либо широких извилин), лиссенцефалии или агирии (сглаженности либо отсутствия извилин), порэнцефалии (наличия патологических кистозных полостей в ткани мозга); агенезия мозолистого тела, расширение ликворных пространств, умеренная гидроцефалия, нарушения миелинизации.
Симптомы микроцефалии
Объем черепа у ребенка с микроцефалией уменьшен уже при рождении, в дальнейшем его развитие заметно отстает от возрастной нормы. Отмечается преобладание лицевого черепа над мозговым. Типичный внешний вид больного с микроцефалией характеризуется узким и скошенным лбом, выступающими надбровными дугами, большими ушами. Большой родничок и черепные швы закрываются уже в первые месяцы жизни. В дальнейшем больные с микроцефалией обычно отстают в массе и росте (вплоть до карликовости), имеют диспропорциональное телосложение, узкое высокое (готическое) небо, большие редкие зубы.
Неврологические нарушения при микроцефалии могут включать мышечную дистонию, спастические парезы, атаксию, судороги, косоглазие. Часто дети с микроцефалией могут страдать эпилепсией и детским церебральным параличом. Дети с микроцефалией поздно начинают держать головку, сидеть, ползать, ходить. Отмечается грубая задержка речевого развития, нечеткость артикуляции, резкая ограниченность словарного запаса, нарушение понимания обращенной речи.
Степень интеллектуальных нарушений у ребенка с микроцефалией может варьировать от дебильности до идиотии. При нерезко выраженной умственной отсталости больные с микроцефалией могут быть обучаемы, способны к самообслуживанию и выполнению несложных поручений. Однако в большинстве случаев дети с микроцефалией требуют ухода, контроля и надзора со стороны взрослых.
По особенностям темперамента дети с микроцефалией могут быть отнесены к торпидной или эретической группе. В первом случае детям свойственна малоподвижность, вялость, безучастность к окружающему, пассивно-подражательная деятельность; во втором случае – гиперактивность, суетливость, подвижность, неустойчивое внимание. Эмоциональная сфера у больных с микроцефалией остается относительно сохранной: дети приветливы, добродушны; реже - эмоционально неустойчивы и склонны к аффективным вспышкам.
Диагностика микроцефалии
Пренатальная диагностика микроцефалии основана на сравнении биометрических параметров плода, получаемых в процессе динамического ультразвукового наблюдения. Однако чувствительность акушерского УЗИ в диагностике микроцефалии составляет всего 67%, а сам порок выявляется только после 27-30 недель беременности. Поэтому при подозрении на микроцефалию, связанную с хромосомной или генетической патологией, УЗИ-скрининг всегда должен дополняться инвазивной пренатальной диагностикой (биопсией хориона, амниоцентезом или кордоцентезом) и кариотипированием плода.
После рождения диагноз микроцефалии подтверждается на основании визуального осмотра новорожденного: уменьшения окружности головы более чем на 2SD-3SD от средней нормы, диспропорции лицевой и мозговой частей черепа. Дети с микроцефалией должны быть проконсультированы генетиком на предмет выявления наследственных заболеваний.
Для определения степени и прогноза микроцефалии важно проведение полного инструментального неврологического обследования: нейросонографии, ЭЭГ, ЭхоЭГ, КТ и МРТ головного мозга. Рентгенография черепа позволяет дифференцировать микроцефалию от краниосиностоза.
Лечение микроцефалии
Патогенетического лечения микроцефалии не существует, поэтому медицинская помощь в основном сводится к симптоматической поддержке больных. На регулярной основе показано проведение медикаментозных курсов, улучшающих обменные процессы в мозговой ткани (пирацетам, пиритинол, витаминные комплексы), по показаниям - противосудорожные и седативные препараты. В рамках реабилитационных мероприятий детям с микроцефалией необходимы занятия лечебной физкультурой, массаж, трудотерапия.
Дети с микроцефалией нуждаются в диспансерном наблюдении педиатра и детского невролога, ежемесячной антропометрии. Воспитание и обучение детей с микроцефалией проводится специалистами дефектологами; коррекция системного недоразвития речи - логопедами. Реабилитационные мероприятия направлены на максимальную адаптацию и социализацию детей с микроцефалией.
Прогноз и профилактика микроцефалии
Прогноз в отношении продолжительности жизни и социализации вариабелен. Некоторые дети способны к обучению в коррекционной школе, овладению элементарными навыками самообслуживания. В целом прогноз при микроцефалии неблагоприятен: продолжительность жизни таких пациентов снижена, большинство из них пожизненно находятся в специнтернатах для умственно-отсталых.
Профилактика микроцефалии у детей предусматривает тщательное планирование беременности, обследование на инфекции (TORCH-комплекс, ПЦР), антенатальную охрану плода. Раннее внутриутробное выявление микроцефалии является основанием для решения вопроса об искусственном прерывании беременности. Медико-генетическое консультирование семей, имеющих детей с микроцефалией, необходимо для оценки потенциального риска при последующих беременностях.
Макроцефалия
Макроцефалия – увеличение размеров головного мозга с сопутствующим увеличением размеров головы. Помимо увеличенного головного мозга, данный синдром может сопровождаться повышением внутричерепного давления и другой неврологической симптоматикой, гипертермией, нарушениями зрения, реже встречается изолированно. Диагностируется на основании измерения размеров черепа, в том числе в динамике, подтверждается результатами КТ и МРТ. Проводится этиотропная терапия заболевания, в клинике которого имеет место макроцефалия. Обязательна дегидратационная терапия, возможно оперативное лечение.
Макроцефалия является синдромом, сопровождающим многие нозологические формы, как врожденные, так и приобретенные. Изначально под данным термином понимали исключительно увеличенный объем мозга, что само по себе может быть вариантом нормы, в последующем критерии патологии сузились. Макроцефалия, как правило, встречается в раннем возрасте, одинаково часто поражает мальчиков и девочек. Диагностика самого синдрома обычно не вызывает сомнений, при этом большинство заболеваний, лежащих в его основе, являются крайне опасными для жизни ребенка. Внутричерепное давление, как правило, нарастает быстрыми темпами, что требует высокой скорости реагирования врачей, срочного выяснения причины развития внутричерепной гипертензии и назначения соответствующей терапии. Этим обусловлена высокая актуальность макроцефалии в педиатрии.
Причины макроцефалии
Так называемая истинная макроцефалия практически не встречается. Данное заболевание является врожденным, причины в настоящее время неизвестны. В современной педиатрии макроцефалия обычно связана с гидроцефалией или рахитом, либо является наследственно детерминированной (семейная макроцефалия). Последний вариант диагностируется крайне редко, течение доброкачественное. Макроцефалия при рахите вызвана остеопорозом и истончением плоских костей черепа.
Повышение внутричерепного давления при гидроцефалии приводит к расширению желудочков мозга избыточной спинномозговой жидкостью, вследствие чего происходит увеличение объема мозга, а затем – головы в целом. Причинами гидроцефалии могут быть внутриутробные инфекции, опухоли, родовая травма новорожденного, редкие сосудистые мальформации и аномалии развития головного мозга, например, аномалия Киари II типа, аномалия Денди-Уокера, стеноз сильвиева водопровода мозга и т. д. Риск развития макроцефалии повышают любые состояния, вызывающие отек мозга у ребенка, включая менингит, сепсис, болезни накопления и многие другие заболевания.
Симптомы макроцефалии
Основной симптом макроцефалии – это увеличенный размер головы. В случае доброкачественной семейной макроцефалии обычно увеличена лобная часть, при рахитической природе состояния отмечается равномерное увеличение всех отделов черепа. При гидроцефалии происходит неравномерное увеличение мозгового черепа, которое может сопровождаться видимым выбуханием большого родничка и сдавлением глазодвигательных нервов. Размер головы при этом не только не соответствует возрастной норме, но и увеличивается в динамике, что всегда может отследить педиатр. Редкие случаи истинной макроцефалии проявляются стойкой гипертермией, головными болями, раздражительностью и рвотой, значительным ухудшением зрения.
Остальные симптомы определяется основным заболеванием. При семейной макроцефалии дополнительные проявления, включая неврологические, отсутствуют. Если макроцефалия имеет место в клинике рахита, можно наблюдать типичную клинику данного заболевания. При гидроцефалии любой этиологии увеличение размера черепа всегда сопровождается признаками повышенного внутричерепного давления: заторможенностью и вялостью ребенка, плохим аппетитом, рвотой, головной болью у детей старшего возраста. Офтальмоскопия позволяет выявить отек зрительного нерва (у младенцев отек может отсутствовать вследствие больших компенсаторных возможностей костей черепа и родничков).
Диагностика макроцефалии
У детей младшего возраста, особенно – до года увеличение размеров черепа выявляется при плановом осмотре педиатра. У детей старшего возраста патология может быть обнаружена родителями. Дифференциальная диагностика разных форм данного синдрома основывается на сопутствующих признаках. Увеличенный размер головы без другой симптоматики свидетельствует в пользу наследственной этиологии. Для подтверждения измеряется объем головы родителей. Для рахита характерны типичные проявления со стороны нервной системы и костного скелета.
Гидроцефалию можно заподозрить при наличии симптомов повышенного внутричерепного давления. Диагноз подтверждается после обнаружения причины гидроцефалии. Для этого чаще всего применяются КТ И МРТ, позволяющие увидеть опухоль, сосудистую мальформацию головного мозга и аномалии развития, способствующие нарушению оттока спинномозговой жидкости. Кроме того, в процессе диагностики может использоваться нейросонография, которая является менее информативной, однако входит в стандартный план обследования детей до года. Данные лабораторных исследований играют решающую роль в случае инфекционной природы увеличения размеров мозга, а также при болезнях накопления.
Лечение и прогноз макроцефалии
Проводится этиотропная терапия патологического процесса, вызвавшего увеличение размеров головы. Для лечения инфекционных заболеваний назначается противовирусная и антибиотикотерапия, при рахите осуществляется специфическая терапия препаратами витамина D. В случае гидроцефалии необходимо устранить причину, вызывающую повышение внутричерепного давления. В качестве временной меры может выполняться шунтирование, но длительное применение данного метода сопряжено с инфекционными осложнениями, поэтому в подобных случаях необходимо скорейшее определение дальнейшей врачебной тактики. Для опухолей и некоторых аномалий возможно только хирургическое лечение. В ряде случаев применяется дегидратационная терапия с использованием диуретиков.
Снизить риск развития синдрома позволяет профилактика возможных внутриутробных факторов риска для плода: травм, инфекций и т. д. Прогноз неблагоприятный за исключением случаев семейной макроцефалии. Даже при успешном излечении высока вероятность сохранения остаточной неврологической симптоматики, отставания в умственном и физическом развитии, симптоматической эпилепсии и других неврологических осложнений.
Читайте также: