Микроскопический полиангиит
Добавил пользователь Алексей Ф. Обновлено: 27.01.2026
Первым термин «Микроскопический узелковый периартериит» использовал F.Wohlwwill в 1922 г. при описании двух пациентов с трансмуральным периартериитом и гломерулонефритом. Оба пациента погибли от болезни, протекавшей с миалгиями, парезом и гломерулонефритом. Ни в одном из случаев не было обнаружено ни макроскопических изменений сосудов, ни аневризм, ни узелков, характерных для классического узелкового периартериита. F.Wohlwwill был убежден, что описанное им заболевание представляет собой форму узелкового периартериита. Он также обнаружил в образцах признаки венулита, но считал, что вовлечение вен, по крайней мере, частично - независимо от артериита. Некоторые авторы еще до F.Wohlwwill (в том числе Оphuls, Beitzke, Walter и von Haun) описывали поражение вен, которое в настоящее время считается более характерным для микроскопического полиангиита, нежели для классического узелкового полиатериита. Микроскопический полиангиит имеет быстропрогрессирующее течение, с развитием осложнений, приводящих к летальному исходу. Причиной смерти обычно являются массивные легочные кровотечения, инфекционные осложнения. Редкость патологии создает дополнительные трудности своевременной диагностики заболевания.
Пациентка Ш, 58 лет, заболела остро в начале декабря 2007 г., когда появились общая слабость, повышение температуры тела до 37-37,5С, заложенность носа, малопродуктивный кашель. Диагностирована ОРВИ, начата терапия ципрофлоксацином 1,0 г/сут, эреспалом. На фоне проводимой терапии сохранялось повышение температуры тела до 38С, потливость. На третьи сутки заболевания выполнена рентгенография органов грудной клетки, выявлена пневмоническая инфильтрация в нижней доле правого легкого. Диагностирована очаговая пневмония, продолжена амбулаторная терапия ципрофлоксацином. Однако, состояние пациентки быстро ухудшалось: нарастала интоксикация, появились кровохарканье, одышка при физической нагрузке, похудание.
Госпитализирована в ЦКБ Гражданской авиации с диагнозом: очаговая пневмония. Анализ крови при поступлении: гемоглобин 82 г/л, лейкоциты 20х109/л, СОЭ 80 мм/час. При КТ грудной клетки 24.12.07 подтвержден диагноз двусторонней полисегментарной пневмонии (рис. 1). Назначен роцефин 2,0 г/сут. Осмотрена фтизиатром, который исключил специфическое поражение легких.
Рис 1. КТ органов грудной клетки с реконструкцией от 24.12.2007: двусторонняя сливная пневмония. Видны зоны инфильтрации легочной ткани.
А – фронтальная плоскость; Б, В – горизонтальная плоскость. Через 2 недели при контрольной рентгенографии легких, а затем при КТ органов грудной клетки от 11.01.08. выявлена отрицательная динамика: тотальная пневмония с элементами деструкции справа, сливная нижнедолевая пневмония слева, реактивный плеврит (рис. 2). С подозрением на тотальную двухстороннюю септическую деструктивную пневмонию 11.01.08 больная была переведена в отделение реанимации центра гнойно-септической хирургии НМХЦ им. Н.И.Пирогова.
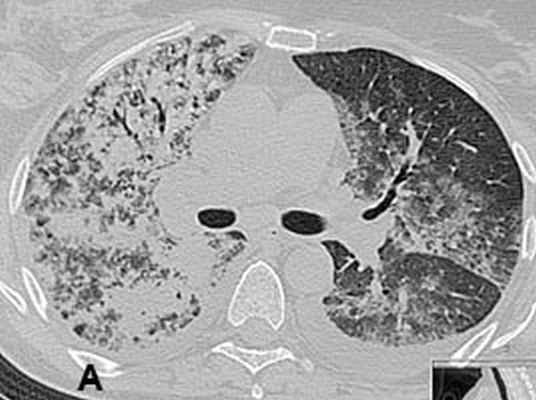 | 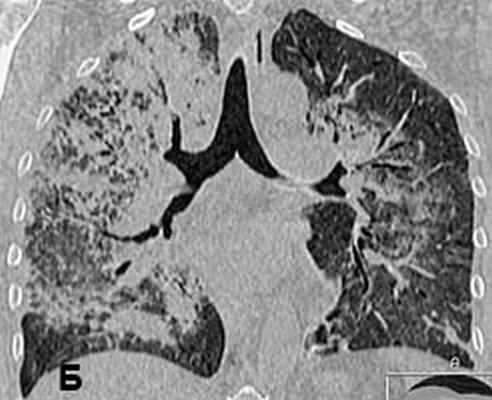 |
Рис 2. КТ органов грудной клетки от 11.01.2008. с реконструкцией: обширная диссеминация мелких «просовидных» теней на фоне диффузной очаговой пневмонии: инфильтрации со склонностью к слиянию; выраженные интерстициальные изменения.
При поступлении состояние тяжелое. Вынужденное положение в кровати с приподнятым головным концом. Температура тела 39,30С. Тахипное до 26-30 в мин. Над всеми отделами легких выслушивались крепитирующие хрипы. Кровохарканье до 80-100 мл. Пульс 118 в мин., ритмичный, удовлетворительного наполнения. Тоны сердца приглушены, АД 140/65 мм рт. ст. Анализ крови от 12.01.08: гемоглобин 80 г/л, лейкоцитоз до 16х109/л, СОЭ 80 мм/час, п – 5%, с – 73%, л - 15%, м - 5%, э – 2%, тромбоциты 460х109/л. АСТ 54 ед/л, АЛТ 46,3 ед/л, билирубин общ. 25,8 мкмоль/л, билирубин пр. 6,1 мкмоль/л, амилаза 118 ед/л, креатинин 91,6 мкмоль/л, белок 63 г/л, альбумин 28 г/л. СРБ – 24 мг/л. Фибриноген 7,88 г/л. Анализ мочи от 12.01.08 – относительная плотность 1013, белок 0.033 г/л, л - 2-4 в п/зр., эр - 17 в п/зр.
У больной заподозрен микроскопический полиангиит. С целью морфологического подтверждения рабочего диагноза через двое суток после поступления в стационар была проведена краевая резекция легкого из миниторакотомного доступа. После этого начата пульс-терапия солумедролом 0.5 г/сутки в течение 4-х дней, однократно циклофосфан 800 мг/сут. в/в капельно. Своеобразная рентгенологическая картина легких в виде обширной диссеминации легких, просовидных теней на фоне диффузной очаговой инфильтрации со склонностью к слиянию и выраженных интрестициальных изменений, развившихся на фоне антибактериальной терапии требовала проведения дифференциальной диагностики между грибковым поражением легких (аспергиллез, криптококкоз), туберкулезом легких, синдромом Гудпасчера.
На основании тотального деструктивного поражения легких с длительно сохранявшимся кровохарканьем, поражением почек, повышением титра АНЦА к протеиназе и данных гистологического исследования - у пациентки диагностирован микроскопический полиангиит. Критерии диагностики заболевания у пациентки полностью соответствовали критериям микроскопического васкулита, предложенных Watts R.A., Scott D.S. (2003): инфильтративно-деструктивное поражение легких (у 50% пациентов), поражение почек (у 100% пациентов), антинейтрофильные антитела (в остром периоде - до 100% пациентов), гистологическое подтверждение.
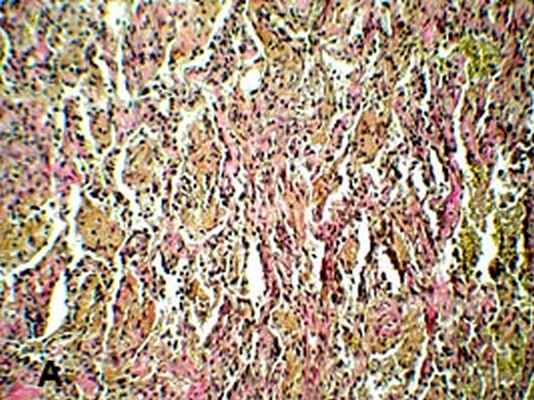 |  |
Рис 3. Биопсия легкого. Микрофото: А - альвеолы со склерозированными перегородками заполнены фибрином; Б - склероз стенок бронхов и артерий.
На фоне проводимой пульс-терапии солумедролом и циклофосфаном состояние и самочувствие быстро улучшались: температура тела стойко нормализовалась на 2 день, кровохарканье сохранялось еще 4 суток. В дальнейшем проводилась терапия метипредом 40 мг/сут., 2 недели аугментином 2,0 г/сут., противогрибковая терапия вифендом, (до иммунологического подтверждения диагноза). Клинический анализ крови, мочи от 7.02.08 в пределах нормы. В удовлетворительном состоянии 8.02.08 выписана на амбулаторное лечение. В марте 2008 приступила к работе. При контрольной КТ органов грудной клетки в марте 2008: инфильтративных изменений в легких не выявлено (рис 4). Клинический анализ крови в пределах нормы. Жалоб нет. Начато снижение дозы метипреда по схеме, к терапии добавлен метотрексат 10 мг/нед. с гормонсберегающей целью.
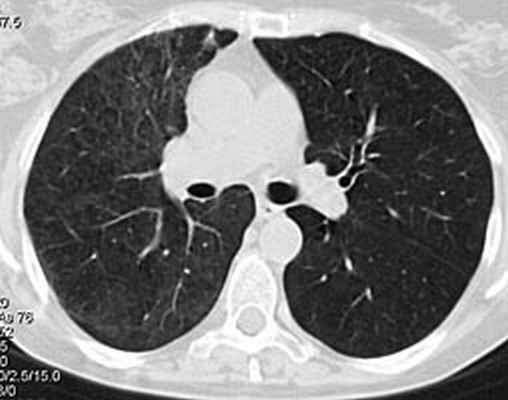 | 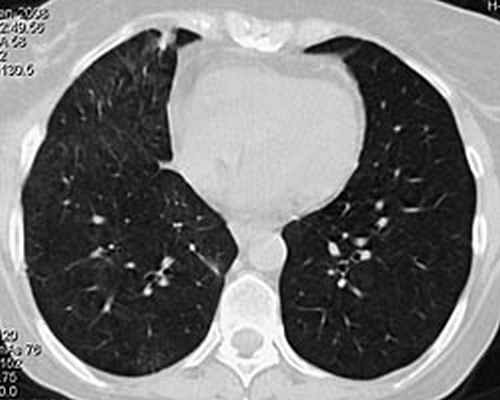 |
Рис 4. КТ органов грудной клетки 18.03. 2008: в легких изменений нет. Особенностью представленного клинического наблюдения явилось деструктивное поражение легких в дебюте заболевания, являющееся проявлением микроскопического полиангиита лишь в 50% случаев. Адекватное, максимально быстрое начало лечения при столь грозном заболевании позволяет достигать ремиссии более чем у 65% пациентов.
© 2022 ФГБУ «НМХЦ им. Н.И. Пирогова» Минздрава России. Использование материалов сайта полностью или частично без письменного разрешения строго запрещено.
Микроскопический полиангиит
Обслуживание на двух языках: русский, английский.
Оставьте свой номер телефона, и мы обязательно перезвоним вам.
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначить только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Статья проверена врачом-ревматологом Филатовой Е.Е., носит общий информационный характер, не заменяет консультацию специалиста.
Для рекомендаций по диагностике и лечению необходима консультация врача.
В Клиническом госпитале на Яузе проводится экспертная диагностика и высокотехнологичное лечение микроскопического полиангиита. Диагностический комплекс включает: лабораторное обследование (в т.ч. иммунологический анализ крови), УЗИ внутренних органов и систем, биопсию тканей в зоне поражения и их гистологическое исследование. Индивидуальная схема лечения включает фармакотерапию и методы экстракорпоральной гемокоррекции.
О развитии микроскопического полиангиита
Микроскопическим полиангиитом (МП) называют заболевание аутоиммунной природы, в основе которого лежит образование аутоантител к клеточным элементам (цитоплазме) собственных нейтрофилов. Это, в свою очередь, приводит к воспалению мелких сосудов (ангииту) и развитию некроза окружающих тканей.
Причина данного заболевания не установлена, однако ученые склоняются к мультифакторной теории возникновения недуга. Отсутствие своевременной адекватной терапии практически со 100% вероятностью приводит к некротическим изменениям в почечной ткани. Развивается гломерулонефрит с быстро прогрессирующей почечной недостаточностью, трудно поддающейся лечению. Когда в процесс вовлечены сосуды бронхолегочной системы, возможно кровохарканье и легочное кровотечение.
Симптомы
На начальном этапе болезни специфические признаки отсутствуют. Пациентов могут беспокоить незначительное недомогание, лихорадка, снижение массы тела, миалгия и артралгия.
Симптомы развернутой клинической картины напрямую зависят от органа, который наиболее поражен недугом:
Дыхательная система страдает в более, чем 50% случаев. У пациентов диагностируют некротический ринит, воспалительные процессы в легких, кровохарканье. Наиболее грозное осложнение в подобной ситуации — легочное кровотечение.
Почки. Микроскопический полиангит поражает почки у 100% больных. Это способствует возникновению гломерулонефрита с прогрессирующей почечной недостаточностью. Оптимальным методом терапии в таком случае считается гемодиализ и последующая трансплантация почки.
Опорно-двигательный аппарат. Не менее половины пациентов отмечают поражение суставов, которое сопровождается нарушением двигательной функции и болями.
Кожа. Для МП характерны кожные проявления: обширные или точечные геморрагические высыпания с элементами некроза, на месте которых образуются язвы.
Нервная система. Больного беспокоят симптомы полинейропатии — локальные изменения кожной чувствительности, «мурашки», покалывания, двигательные нарушения, зябкость, потливость рук,.
Нарушения зрения, эписклерит или кератоконъюнктивит встречаются у 25% пациентов.
Диагностика микроскопического полиангиита в Клиническом госпитале на Яузе
Лабораторные исследования
- Общий анализ крови.
- Общий анализ мочи.
- Подсчет количества тромбоцитов.
- Иммунологический анализ (определение антинейтрофильных цитоплазматических антител — pANCA).
Инструментальное обследование
- УЗИ мочеточников и обеих почек.
- Допплерография сосудов.
- Рентгенография легких.
- Биопсия тканей пораженного органа и гистологический анализ полученного материала.
С уверенностью можно говорить о микроскопическом полиангиите, если у пациента присутствуют следующие симптомы:
- Периодическая лихорадка, снижение массы тела, общее недомогание.
- Наличие инфильтратов в легких и сопутствующее кровохарканье.
- Активно прогрессирующий гломеролунефрит.
- Жалобы на миалгии и воспаление суставов.
- Поражение кожи в виде геморрагических высыпаний.
- Высокий уровень pANCA и антимиелопероксидазы в крови.
Лечение
В Клиническом госпитале на Яузе схема лечения микроскопического полиангиита разрабатывается индивидуально для каждого пациента с учётом результатов обследования и общего состояния больного. Основным средствами для лечения МП считаются цитостатики и глюкокортикоидные гормоны.
В особо тяжелых случаях может быть рекомендован гемодиализ и трансплантация почки.
Улучшить состояние пациента, снизить активность патологического процесса, нашим специалистам помогают методы экстракорпоральной гемокоррекции.
Экстракорпоральная гемокоррекция при микроскопическом полиангиите
Применение ЭГ при микроскопическом полиангиите преследует следующие цели:
- улучшить восприимчивость организма к медикаментозной терапии, что позволит максимально снизить дозировки лекарств и уменьшить вероятность развития побочных эффектов,
- уменьшить уровень pANCA и антимиелопероксидазы в крови,
- нивелировать внешние проявления болезни (сыпь),
- восстановить функцию почек и других систем,
- повысить качество жизни и замедлить инвалидизацию пациента
В Клиническом госпитале на Яузе используются следующие методы ЭГ:
- криоаферез;
- каскадная фильтрация плазмы;
- иммуносорбция;
- высокообъемный плазмообмен;
- экстракорпоральная фармакотерапия;
- фотоферез.
Показаниями к проведению экстракорпоральной гемокоррекции служат поражение почек и дыхательной системы при МА, нарастание признаков почечной недостаточности, отрицательная динамика лабораторных показателей, некроз кожи в зоне высыпаний, появление кровохарканья.
При подозрении на микроскопический полиангиит запишитесь на прием к ревматологу Клинического госпиталя на Яузе, чтобы взять заболевание под контроль и снизить риск грозных осложнений.
Цены на услуги Вы можете посмотреть в прайсе или уточнить по телефону, указанному на сайте.
Цены на услуги ревматолога
- Стандартные консультации
- Прием (осмотр, консультация) врача-ревматолога первичный 3 900 руб.
- Прием (осмотр, консультация) врача-ревматолога повторный 3 600 руб.
- Консультации экспертов
- Прием (осмотр, консультация) врача-ревматолога Бородина О.О. 7 000 руб.
- Телеконсультации
- Удаленная консультация врача-ревматолога первичная 2 500 руб.
- Удаленная консультация врача-ревматолога повторная 2 500 руб.
- Эндоскопия
- Капсульная эндоскопия 50 000 руб.
Внимание! Цены на сайте могут отличаться.
Пожалуйста, уточняйте актуальную стоимость у администраторов по телефону.Наши специалисты:
![Бородин Олег Олегович]()
БородинОлег Олегович Врач-ревматолог
Прием пациентов с 16 лет
Стоимость приема: 7000 ₽Микроскопический полиангиит: особенности течения и трудности диагностики (клиническое наблюдение)
Микроскопический полиангиит (МПА) — это вариант узелкового полиартериита, протекающий с поражением мелких сосудов, ассоциированный с антинейтрофильными цитоплазматическими антителами (АНЦА) к протеиназе-3 и миелопероксидазе.
В статье приводится клинический случай развития МПА у женщины 58 лет. В течение года пациентка неоднократно была госпитализирована. Заболевание протекало с преимущественным поражением почек по типу быстропрогрессирующего гломерулонефрита при недостаточной выраженности клинических проявлений со стороны других органов и систем. Диагностический поиск включал: злокачественные новообразования, инфекции, гинекологическую, гематологическую, хирургическую патологию. На основании данных лабораторных иммунологических исследований — повышения титра АНЦА к протеиназе-3 — было заподозрено системное заболевание. Диагноз был подтвержден гистологически. Для достижения контроля над процессом потребовалось применение иммуносупрессивной терапии.
Случай иллюстрирует сложность своевременной верификации диагноза и выбора эффективного лечения в связи с полиморфизмом клинической картины и нетипичностью проявлений МПА.Ключевые слова: микроскопический полиангиит, системный васкулит, АНЦА, лихорадка, иммуносупрессивная терапия, склерозирующий гломерулонефрит, нефробиопсия.
Utkina E.I. 1 , Myasoedova S.E. 1 , Manokhin V.Yu. 2 , Afanasyeva I.P. 2 , Urusova E.V. 2
1 Ivanovo State Medical Academy
2 Ivanovo Regional Clinical HospitalMicroscopic polyangiitis (МРА) is a variant of nodular polyarteritis, which proceeds with the lesion of small vessels, associated with antineutrophil cytoplasmic antibodies (ANCA) to proteinase-3 and myeloperoxidase. The article presents a clinical case of the development of МРА in a woman of 58 years. The patient was hospitalized to the city hospitals many times throughout the year. The disease proceeded with the renal injury by the type of rapidly progressive glomerulonephritis with minimal symptoms from other internal organs and systems. Diagnostic search included: cancer, infections, gynecological, hematological, surgical pathology. Based on the data of laboratory immunological studies — an increase in the ANCA titer to proteinase-3 — a systemic disease was suspected. The diagnosis was confirmed histologically. To achieve control of the process, the use of immunosuppressive therapy was required.
The case illustrates the complexity of timely verification of the diagnosis and the choice of effective treatment in connection with polymorphism of the clinical picture and atypical manifestations of microscopic polyangiitis.Key words: microscopic polyangiitis, systemic vasculitis, ANCA, fever, immunosuppressive therapy, sclerosing glomerulo-nephritis, kidney biopsy.
For citation: Utkina E.I., Myasoedova S.E., Manokhin V.Yu. et al. Microscopic polyangiitis: peculiarities of a clinical course and difficulties of diagnostics (case report) // RMJ. Medical Review. 2018. № 1(II). P. 118–120.В статье рассмотрены особенности течения и трудности диагностики микроскопического полиангиита. Приводится клинический случай развития микроскопического полиангиита.
В последние годы отмечена тенденция к росту распространенности системных васкулитов (СВ) — заболеваний, основным морфологическим признаком которых является воспаление сосудистой стенки, а клинические проявления определяются типом, калибром, локализацией пораженных сосудов и тяжестью иммуновоспалительных изменений [1]. Несмотря на внедрение в медицинскую практику современных лабораторных и инструментальных методов, данная категория нозологий чаще всего вызывает затруднение в верификации и постановке диагноза, что связано с полиморфизмом клинической картины, смазанностью и нетипичностью проявлений, особенно в дебюте заболевания, и требует обширного дифференциально-диагностического поиска. Между тем без своевременной патогенетической терапии СВ прогрессирует, нередко с развитием трагических осложнений.
Одним из труднодиагностируемых СВ является микроскопический полиангиит (МПА) — некротизирующий васкулит преимущественно мелких сосудов (капилляров, венул, артериол) с отсутствием иммунных депозитов [2]. Эта форма СВ была выделена как отдельный вариант узелкового полиартериита J. Davson et al. в 1948 г. ввиду особенностей клинической картины, морфологических и иммунных нарушений. В настоящее время МПА регистрируют почти в 10 раз чаще узелкового полиартериита. Данный васкулит, как правило, развивается в возрасте 55–75 лет, практически с одинаковой частотой у мужчин и женщин, без национальной предрасположенности [3, 4]. МПА относится к группе васкулитов, ассоциированных с антинейтрофильными цитоплазматическими антителами (АНЦА). Это гетерогенная популяция аутоантител, реагирующих с антигенами первичных гранул нейтрофилов и лизосом моноцитов, а именно с ферментами протеиназой-3 (цитоплазматические с-АНЦА) и миелопероксидазой (перинуклеарные р-АНЦА), что приводит к дегрануляции и окислительному взрыву нейтрофилов. Центральное значение в развитии сосудистого воспаления имеет стимуляция адгезии нейтрофилов к эндотелию с его повреждением и развитием микротромбозов, гиперкоагуляции. При этом отмечено, что МПА в 2 раза чаще ассоциирован с р-АНЦА [2, 4–6]. Этиология МПА неизвестна. Установлена определенная связь с генетическими факторами: полиморфизм генов, кодирующих синтез протеиназы-3 и ее основного ингибитора альфа1-антитрипсина (SERPIN A1) предрасполагает к гиперпродукции антител к протеиназе-3 [2]. МПА носит генерализованный характер. Поражение почек отмечается у 90% пациентов и нередко характеризуется быстропрогрессирующим течением с развитием почечной недостаточности, особенно тяжело протекая при наличии антител к протеиназе-3 [7], реже процесс ограничивается клиникой нефротического, остронефритического синдромов, бессимптомной протеинурии, гематурии [2, 8]. Морфологическая картина характеризуется фибриноидным некрозом капилляров клубочков и артериол, экстракапиллярной пролиферацией с эпителиальными или фибринозно-клеточными полулуниями при отсутствии иммунных депозитов в ткани почки [2]. Признаки поражения легких наблюдаются в 30–50% случаев, проявляются кашлем, одышкой, кровохарканьем. При биопсии в легочной ткани обнаруживают некротизирующий альвеолит и септальный капиллярит с нейтрофильной инфильтрацией, пневмофиброз [4, 9]. Поражение кожи (70%) характеризуется геморрагическими или язвенно-геморрагическими высыпаниями преимущественно на коже конечностей, реже livedo reticularis, некрозами кожи и подлежащих мягких тканей [7]. В симптомокомплекс заболевания входят артриты, миалгии, абдоминалгии, общая слабость. Реже встречаются контактная кровоточивость, периферическая ишемическая полинейропатия, энцефалопатия, поражение глаз и носа, стриктуры мочеточников. Диагноз основывается на данных клинической картины, иммунологического и морфологического исследований.Клинический случай
Пациентка П., 58 лет, пенсионерка, без вредных привычек, считает себя больной с конца 2015 г., когда ее стали беспокоить слабость, отсутствие аппетита, немотивированное похудание, периодические подъемы артериального давления (АД) до 190/110 мм рт. ст. При обследовании в поликлинике по месту жительства выявлено повышение скорости оседания эритроцитов (СОЭ) до 40 мм/ч, анемия легкой степени. В январе 2016 г. самостоятельно лечилась антибиотиками по поводу катаральных явлений, повышения температуры тела до субфебрильных цифр — без эффекта. Была госпитализирована в терапевтическое отделение ГКБ № 3 с основным диагнозом: «Хронический бронхит, обострение. Железодефицитная анемия легкой степени». Однако вскоре пациентка была переведена в инфекционное отделение ГКБ № 1 с подозрением на энтеровирусную инфекцию: больную стали беспокоить тошнота, умеренные боли в животе, диарея, сохранялась лихорадка, потеря массы тела. В ходе лечения на фоне внутривенного введения дексаметазона 4 мг самочувствие значительно улучшилось, нормализовалась температура тела и восстановился аппетит.
После выписки из стационара общее состояние вновь стало ухудшаться: нарастала слабость, потеря аппетита, субфебрилитет по вечерам. Больная самостоятельно принимала жаропонижающие препараты с временным эффектом. Спустя 2 мес. присоединились артриты обоих голеностопных суставов с выраженным болевым синдромом, синовитом. Симптоматически лечилась обезболивающими препаратами. К июлю суставной синдром купировался, но стала нарастать слабость, потеря массы тела составила 17 кг, лихорадка достигла фебрильного уровня, периодически беспокоили абдоминалгии. Больная вновь обследовалась амбулаторно: впервые был выявлен мочевой синдром, прогрессировала анемия. Повторно госпитализирована в ГКБ № 1 с диагнозом: «Анемия железодефицитная, средней тяжести, декомпенсация. Лихорадка неясного генеза». Проводился онкопоиск: рентгенография органов грудной клетки — без патологии, фиброэзофагогастродуоденоскопия — поверхностный гастродуоденит, фиброколоноскопия — хронический колит. При бактериологическом посеве кала и исследовании крови на стерильность роста патологической микрофлоры не выявлено. Рентгенологически были определены костно-деструктивные изменения костей таза и черепа. С подозрением на множественную миелому пациентка была осмотрена гематологом, диагноз отвергнут на основании нормальных данных стернальной пункции. Лечилась симптоматически, с неполным эффектом.
В июле 2016 г. с жалобами на интенсивные боли в животе, повышение температуры тела до 38 °С, выраженную общую слабость больная поступила в экстренном порядке в хирургическое отделение ГКБ № 4. При обследовании: гемоглобин 88 г/л, лейкоцитоз (12,8 г/л), лимфопения, тромбоцитоз, СОЭ 70 мм/ч, С-реактивный белок (СРБ) 193,4 мг/л. Динамика лабораторных показателей в течение недели: нарастание креатинина сыворотки до 830 мкмоль/л, мочевины — до 46 ммоль/л. Скорость клубочковой фильтрации 8,13 мл/мин. Анализы мочи: эритроцитурия, протеинурия до 0,66 г/л, ацетон отрицательный. Ультразвуковое исследование (УЗИ) внутренних органов: диффузные изменения в печени и почках, узловые изменения в щитовидной железе; сцинтиграфия костей — без очаговых изменений. Эхо-кардиография: снижение глобальной сократительной функции миокарда. Несмотря на проводимую антибактериальную, дезинтоксикационную, антикоагулянтную терапию, состояние больной ухудшалось: нарастали общая слабость, отвращение к пище, сердцебиение; на плечах и предплечьях появились болезненные подкожные инфильтраты бурого цвета, развился артрит левого плечевого сустава. Переведена в палату интенсивной терапии. Ввиду онконастороженности больная консультирована гинекологом, офтальмологом, неврологом, урологом, повторно инфекционистом: данных за специфическую патологию не выявлено. Осмотрена нефрологом: установлена почечная недостаточность неуточненной этиологии. Первичная почечная патология была исключена, заподозрен паранеопластический синдром. Пациентка была переведена в нефрологическое отделение Ивановской областной клинической больницы (ОКБ) для проведения гемодиализа по экстренным показаниям.
Проведено: консультации специалистов: сосудистый хирург — флебит подкожных вен правого и левого плеча, паравазальный инфильтрат; невролог — энцефаломиелополинейропатия смешанного генеза. Проведена мультиспиральная компьютерная томография (МСКТ) грудной, брюшной полостей, малого таза: объемных образований не выявлено. Лабораторные исследования: повышение острофазовых показателей крови (СОЭ 65 мм/ч, СРБ 29,4 мг/л), ферритин 1687 нг/мл, паратгормон 233,1 пг/мл, положительный ревматоидный фактор (РФ) 20,2 МЕ/мл. Заподозрен недифференцированный системный васкулит. Пациентка была переведена в ревматологическое отделение ОКБ с основным диагнозом: «Недифференцированный системный васкулит с поражением суставов (артриты по анамнезу), почек с исходом в острую почечную недостаточность. Флебиты подкожных вен правого и левого плеча. Анемия смешанного генеза. Энцефаломиелополинейропатия смешанного генеза на фоне васкулита». При углубленном иммунологическом исследовании выявлен умеренно повышенный уровень антинейтрофильных антител к протеиназе-3; антитела к миелопероксидазе не обнаружены, онкомаркеры и антинуклеарный фактор — в пределах референсных значений. К лечению добавлен метилпреднизолон 24 мг/сут, проведен сеанс терапии циклофосфамидом 400 мг внутривенно, противоанемическая терапия. На фоне лечения в течение недели общее самочувствие улучшилось, снизилась температура тела, гемоглобин достиг целевых значений — 127 г/л, нормализовались острофазовые показатели крови и уменьшился уровень азотистых шлаков: креатинин 411 мкмоль/л, мочевина 19,7 ммоль/л. Скорость клубочковой фильтрации соответствовала V стадии хронической болезни почек — 11,2 мл/мин. Заместительная почечная терапия больше не проводилась. Больная продолжила получать иммуносупрессивную терапию, на фоне чего чувствовала себя хорошо.
С целью уточнения диагноза в ноябре 2016 г. пациентка была направлена на обследование в Клинику нефрологии, внутренних и профессиональных болезней им. Е.М. Тареева. При контрольном обследовании: гемоглобин 122 г/л, СОЭ 12 мм/ч, креатинин крови 422 мкмоль/л. При иммунологическом исследовании с-АНЦА к протеиназе-3 снизились до нормальных значений; р-АНЦА к миелопероксидазе, РФ, СРБ, антитела к двуспиральной ДНК, антинуклеарный фактор, криоглобулины, криофибриноген, волчаночный антикоагулянт — не выявлены. На КТ легких обнаружен участок пластинчатого фиброза в 4-м сегменте справа, дистрофически-дегенеративные изменения позвоночника. КТ головного мозга: очаги отека белого вещества левой теменной доли — ишемия или воспаление, вероятно, в рамках васкулита. УЗИ органов брюшной полости и почек: без отрицательной динамики. Выполнена нефробиопсия. При световой микроскопии в гистологическом препарате обнаружено множество склерозированных клубочков — участки сегментарного склероза с изменением петель по типу постнекротического рубцевания с образованием сегментарных фиброзных полулуний, остальные клубочки ишемизированы. Пролиферативных изменений нет. Диффузно-очаговый фиброз интерстиция и атрофия канальцев занимают более 50% паренхимы. Сохранные канальцы гипертрофированы. Диффузно-очаговая неспецифическая интерстициальная инфильтрация мононуклеарами в зонах склероза. Артерии и артериолы — без особенностей. При иммунофлюоресценции IgG, IgA, IgM, C3, C1q, каппа-, лямбда-фрагменты не обнаружены. Заключение по биоптату: диффузный склерозирующий гломерулонефрит с 45% фиброзных полулуний. Выявленная гистологическая картина ткани почки была расценена как характерная для поздней стадии поражения почек при АНЦА-ассоциированном васкулите по типу МПА. При выписке рекомендовалось продолжить иммуносупрессивную терапию, а в случае прогрессирования почечной недостаточности начать лечение программным гемодиализом.
В настоящее время пациентка получает индукционную терапию циклофосфамидом и метилпреднизолоном с положительным эффектом: креатинин сыворотки стабилизировался на уровне 400–450 мкмоль/л, острофазовые и иммунологические показатели крови в пределах нормальных значений.Заключение
Особенностью течения МПА в рассмотренном случае явилось развитие заболевания с преимущественным поражением почек по типу быстропрогрессирующего экстракапиллярного гломерулонефрита при недостаточной выраженности клинических проявлений со стороны других органов и систем, а также наличие в дебюте повышенного титра АНЦА к протеиназе-3 при отсутствии более характерных для МПА антител к миелопероксидазе нейтрофилов. От правильности диагноза и точного определения степени активности процесса зависел выбор, объем и длительность терапии, а также прогноз и возможность дальнейшего ведения больной без заместительной почечной терапии. Диагноз был установлен по результатам нефробиопсии: при гистологическом исследовании подтверждено тяжелое поражение почек (склеротический класс гломерулонефрита) в рамках АНЦА-ассоциированного васкулита — МПА. Также выявлено вовлечение в патологический процесс центральной нервной системы (очаговые изменения вещества мозга в рамках васкулита) и легочной ткани в виде фиброза. Таким образом, на верификацию диагноза потребовалось около года от начала заболевания. Иммуносупрессивная терапия позволила стабилизировать состояние больной и предотвратить прогрессирование заболевания.
В «РМЖ. Медицинское обозрение» №1(I), 2018 была допущена опечатка в номере
свидетельства о регистрации средства массовой информации. Верный номер –
ПИ № ФС77-53569.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.Микроскопический полиангиит: современные представления и возможности терапии
В статье обобщены современные представления о микроскопическом полиангиите (МПА), заболевании из группы системных васкулитов, ассоциированных с антинейтрофильными цитоплазматическими антителами (АНЦА). В основе МПА лежит некротизирующий АНЦА-ассоциированный васкулит без иммунных депозитов, с преимущественным поражением мелких сосудов и без гранулематозного воспаления. Для МПА типичны развитие некротизирующего гломерулонефрита и частое присоединение геморрагического альвеолита, а также нередко отмечается быстропрогрессирующее течение. Обсуждаются современные возможности терапии МПА, прежде всего анти-В-клеточная терапия ритуксимабом.
Ключевые слова
Об авторах
Муркамилов Илхом Торобекович
720020, Кыргызстан, Бишкек, ул. Ахунбаева, 92
720040, Кыргызстан, Бишкек, ул. Тоголок Молдо, 3
ФГАОУ ВО «Первый Московский государственный медицинский университет имени И.М. Сеченова» Минздрава России (Сеченовский Университет)
Россия119991, Российская Федерация, Москва, ул. Трубецкая, 8, стр. 2
720020, Кыргызстан, Бишкек, ул. Ахунбаева, 92
723500, Кыргызстан, Ош, ул. Ленина, 331
720000, Кыргызстан, Бишкек, ул. Киевская, 44
115522, Российская Федерация, Москва, Каширское шоссе, 34а
Список литературы
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11. doi: 10.1002/art.37715
2. Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. 2016;75(9):1583-1594. doi: 10.1136/annrheumdis-2016-209133
3. Бекетова ТВ. Микроскопический полиангиит, ассоциированный с антинейтрофильными цитоплазматическими антителами: особенности клинического течения. Терапевтический архив. 2015;87(5):33-46.
4. Nguyen Y, Pagnoux C, Karras A, Quéméneur T, Maurier F, Hamidou M, et al. Microscopic polyangiitis: Clinical characteristics and long-term outcomes of 378 patients from the French Vasculitis Study Group Registry. J. Autoimmun. 2020;112:102467. doi: 10.1016/j.jaut.2020.102467
5. Wohlwill F. Über die nur mikroskopisch erkennbare Form der Periarteritis nodosa. Virchows Arch Pathol Anat Physiol. 1923;246:36.
6. Wainwright J, Davson J. The renal appearances in the microscopic form of periarteritis nodosa. J Pathol Bacteriol. 1950;62(2):189-196. doi: 10.1002/path.1700620206
8. Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: Presentation, pathology and prognosis. Q J Med. 1985;56(220):467-483.
9. van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet. 1985;1(8426):425-429. doi: 10.1016/s0140-6736(85)91147-x
10. Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: immunodiagnostic and pathogenetic aspects. Clin Exp Immunol. 1993;91(1):1-12. doi: 10.1111/j.1365-2249.1993.tb03345.x
11. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an International Consensus Conference. Arthritis Rheum. 1994;37(2):187-192. doi: 10.1002/art.1780370206
12. Семенкова ЕН, Бекетова ТВ, Коган ЕА, Козловская ЛВ, Мухин НА. Современные представления о микроскопическом полиангиите. Терапевтический архив. 1995;67(5):39-41.
13. Watts R, Lane S, Hanslik T, Hauser T, Hellmich B, Koldingsnes W, et al. Development and validation of a consensus methodology for the classification of the ANCA associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007;66(2):222-227. doi: 10.1136/ard.2006.054593
14. Chung SA, Seo P. Microscopic polyangiitis. Rheum Dis Clin North Am. 2010;36(3):545-558. doi: 10.1016/j.rdc.2010.04.003
18. Shi J, Shen Q, Chen XM, Du XG. Clinical characteristics and outcomes in microscopic polyangiitis patients with renal involvement: a study of 124 Chinese patients. BMC Nephrol. 2019;20(1):339. doi: 10.1186/s12882-019-1535-3
19. Sugiyama K, Sada KE, Kurosawa M, Wada J, Makino H. Current status of the treatment of microscopic polyangiitis and granulomatosis with polyangiitis in Japan. Clin Exp Nephrol. 2013;17(1):51-58. doi: 10.1007/s10157-012-0651-1
20. Lane SE, Watts R, Scott DG. Epidemiology of systemic vasculitis. Curr Rheumatol Rep. 2005;7(4):270-275. doi: 10.1007/s11926-005-0036-5
21. Comarmond C, Crestani B, Tazi A, Hervier B, Adam- Marchand S, Nunes H, et al. Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: A series of 49 patients and review of the literature. Medicine (Baltimore). 2014;93(24):340-349. doi: 10.1097/MD.0000000000000217
22. Бекетова ТВ. Алгоритм диагностики системных васкулитов, ассоциированных с антинейтрофильными цитоплазматическими антителами. Терапевтический архив. 2018;90(5):13-21.
23. Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically distinct subsets within ANCA-associated vascuonline litis. N Engl J Med. 2012;367(3):214-223. doi: 10.1056/NEJMoa1108735
24. Tsuchiya N, Kobayashi S, Hashimoto H, Ozaki S, Tokunaga K. Association of HLA-DRB1*0901-DQB1*0303 haplotype with microscopic polyangiitis in Japanese. Genes Immun. 2006;7(1):81-84. doi: 10.1038/sj.gene.6364262
25. Rahmattulla C, Mooyaart AL, van Hooven D, Schoones JW, Bruijn JA, Dekkers OM, et al. Genetic variants in ANCAassociated vasculitis: A meta-analysis. Ann Rheum Dis. 2016;75(9):1687-1692. doi: 10.1136/annrheumdis-2015-207601
26. Первакова МЮ, Чудинов АЛ, Лапин СВ, Беляева ИБ, Мазуров ВИ, Блинова ТВ, и др. Диагностическая и клиническая значимость определения фенотипа α-1-антитрипсина при системных васкулитах. Научно-практическая ревматология. 2017;55(2):164-168.
27. Hagen EC, Ballieux BE, van Es LA, Daha MR, van der Woude FJ. Antineutrophil cytoplasmic autoantibodies: A review of the antigens involved, the assays, and the clinical and possible pathogenetic consequences. Blood. 1993;81(8):1996-2002.
28. Kallenberg CG, Brouwer E, Weening JJ, Tervaert JW. Antineutrophil cytoplasmic antibodies: Current diagnostic and pathophysiological potential. Kidney Int. 1994;46(1):1-15. doi: 10.1038/ki.1994.239
30. Witko-Sarsat V, Daniel S, Noël LH, Mouthon L. Neutrophils and B lymphocytes in ANCA-associated vasculitis. APMIS Suppl. 2009;127:27-31. doi: 10.1111/j.1600-0463.2009.02473.x
31. Taekema-Roelvink MEJ, Kooten CV, Kooij SV, Heemskerk E, Daha MR. Proteinase 3 enhances endothelial monocyte chemoattractant protein-1 production and induces increased adhesion of neutrophils to endothelial cells by upregulating intercellular cell adhesion molecule-1. J Am Soc Nephrol. 2001;12(5):932-940. doi: 10.1681/ASN.V125932
33. Foucher P, Heeringa P, Petersen AH, Huitema MG, Brouwer E, Tervaert JW, et al. Antimyeloperoxidase-associated lung disease. An experimental model. Am J Respir Crit Care Med. 1999;160(3):987-994. doi: 10.1164/ajrccm.160.3.9807139
34. Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Maeda N, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167(1):39-45. doi: 10.1016/S0002-9440(10)62951-3
35. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20(2):289-298. doi: 10.1681/ASN.2008050497
36. Hao J, Meng LQ, Xu PC, Chen M, Zhao MH. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLoS ONE. 2012;7(5):e38317. doi: 10.1371/journal.pone.0038317
37. Antovic A, Mobarrez F, Manojlovic M, Soutari N, De Porta Baggemar V, Nordin A, et al. Microparticles expressing myeloperoxidase and complement C3a and C5a as markers of renal involvement in antineutrophil cytoplasmic antibody-associated vasculitis. J Rheumatol. 2020;47(5):714-721. doi: 10.3899/jrheum.181347
39. Hu P, Su H, Xiao H, Gou SJ, Herrera CA, Alba MA, et al. Kinin B1 receptor is important in the pathogenesis of myeloperoxidase-specific ANCA GN. J Am Soc Nephrol.2020;31:297-307. doi: 10.1681/ASN.2019010032
41. Щеголева ЕМ, Буланов НМ, Виноградова ЕС, Зыкова АС, Новиков ПИ, Моисеев СВ. Варианты течения и исходы микроскопического полиангиита. Клиническая фармакология и терапия. 2018;27(3):35-40.
42. Hirayama K, Kobayashi M, Usui J, Arimura Y, Sugiyama H, Nitta K, et al. Pulmonary involvements of antineutrophil cytoplasmic autoantibody-associated renal vasculitis in Japan. Nephrol Dial Transplant. 2015;30(Suppl 1):i83-i93. doi: 10.1093/ndt/gfu385
43. Nada AK, Torres VE, Ryu JH, Lie JT, Holley KE. Pulmonary fibrosis as an unusual clinical manifestation of a pulmonary-renal vasculitis in elderly patients. Mayo Clin Proc. 1990;65(6):847-856. doi: 10.1016/s0025-6196(12)62575-0
46. Ahn JK, Hwang JW, Lee J, Jeon CH, Cha HS, Koh EM. Clinical features and outcome of microscopic polyangiitis under a new consensus algorithm of ANCA-associated vasculitides in Korea. Rheumatol Int. 2012;32(10):2979-2986. doi: 10.1007/s00296-011-2079-4
47. Arienti F, Franco G, Monfrini E, Santaniello A, Bresolin N, Saetti MC, et al. Microscopic polyangiitis with selective involvement of Central and Peripheral Nervous System: A case report. Front Neurol. 2020;11:269. doi: 10.3389/fneur.2020.00269
48. Tauseef A, Asghar MS, Amir M, Zafar M, Anum A, Alvi H, et al. Microscopic polyangiitis: An incidental finding in a patient with stroke. J Community Hosp Intern Med Perspect. 2020;10(1):50-54. doi: 10.1080/20009666.2020.1718479
49. Щеголева ЕМ, Зыкова АС, Буланов НМ, Новиков ПИ, Моисеев СВ, Мухин НА. Современные подходы к диагностике и лечению микроcкопического полиангиита. Клиническая медицина. 2018;96(1):66-72.
50. Фролова НФ, Корсакова ЛВ, Столяревич ЕС, Никонорова НО, Бекетова ТВ. Дебют АНЦА- ассоциированного системного васкулита под маской ревматоидного артрита. Научно-практическая ревматология. 2015;53(6):653-656.
51. Pagnoux C, Seror R, Bérezné A, Rouabhia S, Goulvestre C, Guillevin L. Remittent non-destructive polysynovitis in P-ANCApositive vasculitis patients with anti-CCP antibodies. Joint Bone Spine. 2010;77(6):604-607. doi: 10.1016/j.jbspin.2010.02.013
52. Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: Clinical and laboratory findings in eighty-five patients. Arthritis Rheum. 1999;42(3):421-430. doi: 10.1002/1529-0131(199904)42:3 3.0.CO;2-6
53. Rodgers H, Guthrie JA, Brownjohn AM, Turney JH. Microscopic polyarteritis: Clinical features and treatment. Postgrad Med J. 1989;65(766):515-518. doi: 10.1136/pgmj.65.766.515
56. Walsh M, Merkel PA, Peh CA, Szpirt WM, Puéchal X, Fujimoto S, et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N Engl J Med.2020;382(7):622-631. doi: 10.1056/NEJMoa1803537
57. Бекетова ТВ, Насонов ЕЛ. Десятилетний опыт индукционной и поддерживающей терапии ритуксимабом у пациентов с АНЦА-ассоциированными системными васкулитами. Современная ревматология. 2020;14(1):12-19.
58. Moiseev S, Novikov P, Bulanov N, Zykova A, Safonova E. Assessing cardiovascular risk in patients with antineutrophil cytoplasmic antibody-associated vasculitis: Comment on the article by Wallace et al. Arthritis Rheumatol. 2020;72(1):200-201. doi: 10.1002/art.41080
Микроскопический полиангиит
Микроскопический полиангиит - генерализованный некротизирующий васкулит, протекающий с преимущественным поражением мелких сосудов (капилляров, артериол, венул) легких, почек и кожи. При микроскопическом полиангиите может отмечаться кашель, одышка, боли в грудной клетке, легочное кровотечение, быстропрогрессирующий гломерулонефрит, почечная недостаточность, кожные петехиальные высыпания, артралгии. Лабораторным подтверждением микроскопического полиангиита служит обнаружение антител к цитоплазме нейтрофилов. Лечение микроскопического полиангиита включает иммуносупрессивную терапию глюкокортикоидами и цитостатиками, плазмаферез; при развитии почечной недостаточности - гемодиализ или трансплантацию почки.
Общие сведения
Микроскопический полиангиит (микроскопический полиартериит) – заболевание из группы системных васкулитов, характеризующееся поражением сосудов мелкого калибра с явлениями некротизирующего гломерулонефрита и легочного капиллярита. В связи с особенностями морфологических, клинических и иммунологических нарушений в 1948 г. микроскопический полиангиит был выделен в отдельную нозологию. Распространенность микроскопического полиангиита составляет 0,36 случаев на 100 тыс. населения. В ревматологии заболевание регистрируется в 10 раз чаще, чем узелковый периартериит и в 2 раза чаще, чем гранулематоз Вегенера. Микроскопический полиангиит практически с одинаковой частотой диагностируется у мужчин и женщин; чаще развивается у лиц старше 50-60 лет.
Причины
Этиология микроскопического полиангиита неизвестна. В настоящее время ведется изучение иммуногенетических аспектов патологии, исследуется пусковая роль вирусной инфекции в развитии заболевания. Вместе с тем, достоверно известно, что основной механизм патогенеза некротизирующего васкулита связан с образованием антител к цитоплазме нейтрофилов (ANCA), вызывающих повреждение эндотелия сосудов. Аутоантитела обладают специфичностью в отношении миелопероксидазы (p-ANCA) и сериновой протеиназы (c-ANCA), реже – других ферментов нейтрофилов. В активную стадию заболевания антинейтрофильные цитоплазматические антитела выявляются у всех пациентов.
Патоморфологические изменения при микроскопическом полиангиите представлены некротизирующим васкулитом мелких сосудов (артериол, венул, капилляров) без явлений гранулематозного воспаления. Наиболее характерно поражение сосудов почек (некротизируюший гломерулонефрит), легких (геморрагический альвеолит, капиллярит) и кожи (лейкоцитокластический венулит). Изредка поражаются артерии среднего и крупного калибра.
Симптомы микроскопического полиангиита
Дебют микроскопического полиангиита характеризуется появлением неспецифических гриппоподобных симптомов: субфебрильной температуры тела, мигрирующих миалгий и артралгий, общей слабости, недомогания, ночной потливости. Примерно у трети больных развивается поражение верхних дыхательных путей (язвенно-некротический или атрофический ринит, синусит, средний отит) и зрительной системы (конъюнктивит, кератит, эписклерит, увеит). Эти изменения обратимы и подвергаются обратному развитию на фоне терапии иммунодепрессантами.
Возможно возникновение стойких артритов мелких и крупных суставов, синовитов пястно-фаланговых и межфаланговых суставов, периферической полинейропатии. Обычно уже в начальной стадии микроскопического полиангиита обнаруживается поражение кожи в виде сосудистой пурпуры, эритемы, ливедо, узелковых или буллезных высыпаний, реже – язвенных изменений и асептических некрозов мягких тканей.
Ведущими проявлениями, определяющими течение и прогноз микроскопического полиангиита, служат легочный и почечный синдромы. Геморрагический альвеолит развивается более чем у половины пациентов. Клинические проявления легочного синдрома включают кашель, прогрессирующую одышку, боль в грудной клетке, нарастающую дыхательную недостаточность. Типично развитие кровохарканья, а иногда – профузного легочного кровотечения, которое является ведущей причиной смерти пациентов с микроскопическим полиангиитом.
Поражение почек по типу некротического гломерулонефрита обнаруживается практически во всех случаях заболевания. Патологический симптомокомплекс характеризуется стойкой протеинурией, гематурией, нефротическим синдромом, мягкой или умеренной артериальной гипертензией. Течение гломерулонефрита при микроскопическом полиангиите быстропрогрессирующее, злокачественное, рано приводящее к развитию острой почечной недостаточности. К нечасто встречающимся висцеральным проявлениям микроскопического полиангиита относятся бронхообструктивный синдром, ишемический энтероколит, протекающий с болями в животе и кишечным кровотечением.
Различают несколько вариантов клинического течения микроскопического полиангиита:
- молниеносный – летальный исход наступает в течение нескольких недель вследствие легочного кровотечения или ОПН;
- подострый – характерны тяжелый нефротический синдром или быстропрогрессирующий гломерулонефрит;
- непрерывно рецидивирующий – обострения наступают каждые 6-12 месяцев; в клиническом течении преобладает неспецифическая симптоматика, гломерулонефрит, кровохарканье;
- латентный – доминируют суставной синдром, гематурия, кровохарканье.
Диагностика
В начальной фазе микроскопического полиангиита пациенты довольно часто обследуются у узких специалистов: отоларинголога, офтальмолога, дерматолога, пульмонолога, фтизиатра, нефролога и др. Однако после исключения изолированных синдромов, диагностикой и лечением заболевания занимается ревматолог.
Специфическим иммунологическим маркером микроскопического полиангиита служит обнаружение в сыворотке крови антител к цитоплазме нейтрофилов. С целью оценки степени активности патологического процесса исследуется общий анализ мочи и крови, фибриноген, С-реактивный белок, электролиты, креатинин, железо сыворотки крови. В рамках морфологической верификации патологии может потребоваться проведение биопсии кожи, слизистой оболочки верхних дыхательных путей, легкого, почки. Рентгенография и КТ легких выявляют двустороннюю очаговую инфильтрацию, при длительном течении микроскопического полиангиита – легочный фиброз. Широко используется ультразвуковая и радиоизотопная диагностика (УЗИ почек, сцинтиграфия легких и др.).
Лечение микроскопического полиангиита
Лечение микроскопического полиангиита преследует цели купирования обострений, достижения и поддержания ремиссии. В острой стадии лечение проводится в стационаре. На этом этапе назначается иммуносупрессивная терапия глюкокортикоидами и циклофосфамидом. Возможен вариант проведения пульс-терапии метилпреднизолоном в сочетании с плазмаферезом. В комплексном лечении быстропрогрессирующих, тяжелых форм микроскопического полиангиита используются и другие методы экстракорпоральной гемокоррекции (криоаферез, каскадная фильтрация плазмы, лимфоцитаферез).
Поддерживающая иммуносупрессивная терапия продолжается даже после достижения полной клинико-лабораторной ремиссии. При нарастании почечной недостаточности проводится гемодиализ; в терминальной стадии ХПН решается вопрос о возможности трансплантации почки.
Прогноз и профилактика
Заболевание имеет относительно неблагоприятный прогноз. На фоне лекарственной терапии 5-летняя выживаемость составляет 65%. Плохими прогностическими факторами в отношении общей выживаемости служат кровохарканье, высокий уровень креатинина крови, олигурия, высокая протеинурия, пожилой возраст. Основными причинами смерти выступают легочное кровотечение, острая дыхательная или почечная недостаточность, инфекционные осложнения. Пациенты с микроскопическим полиангиитом нуждаются в длительной терапии иммунодепрессантами и пожизненном наблюдении ревматолога. Профилактика заболевания не разработана.
Читайте также:
