Морфология гиперинсулинизма. Патологическая анатомия инсуломы
Добавил пользователь Владимир З. Обновлено: 29.01.2026
Представлено клиническое наблюдение инсулиномы у девочки 16 лет, у которой отмечены жалобы на приступы потери сознания с последующей ретроградной амнезией, сопровождавшиеся слабостью и головокружением. Больная был переведена в эндокринологическое отделение, где при регулярном контроле гликемии зафиксированы низкие значения (2,0–2,5 ммоль/л). Топический диагноз инсулиномы был установлен с помощью лучевых методов – мультиспиральной компьютерной томографии с контрастным усилением и магнитно-резонансной томографии, при которых в хвосте поджелудочной железы выявлена овальной формы опухоль размерами 12 × 10 мм. Девочке было проведено молекулярно-генетическое исследование гена MEN1 – мутаций в исследованном гене не выявлено. На период предоперационной подготовки с целью профилактики гипогликемического синдрома был назначен прогликем (диазоксид) в дозе 100 мг/сут, на фоне которого отмечалась стабилизация показателей гликемии. В дальнейшем была выполнена лапароскопическая дистальная резекция поджелудочной железы с опухолью. После операции достигнута стойкая эугликемия. При микроскопическом и иммуногистохимическом исследовании подтверждена нейроэндокринная опухоль поджелудочной железы G1. В обсуждении представлены данные об эпидемиологии, диагностике и хирургическом лечении инсулином у детей и взрослых.
Ключевые слова
Для цитирования:
For citation:
Инсулинома – нейроэндокринная инсулинсекретирующая опухоль, возникающая из бета-клеток островков поджелудочной железы (ПЖ). Частота встречаемости инсулином составляет приблизительно 4 случая на 1 млн в год [1, 2]. Инсулинома может возникать спорадически (90%) или являться частью синдрома MЭН1 – множественных эндокринных неоплазий (10%). Спорадические инсулиномы встречаются в возрасте от 8 до 82 лет (средний возраст – 45–50 лет), тогда как инсулиномы, ассоциированные с синдромом MЭН1, поражают более ранний возраст (средний возраст – 25 лет и младше) [3].
У детей инсулиномы встречаются крайне редко, и их точная частота в детской популяции не определена [1, 4]. Средний возраст постановки диагноза составляет 10 лет, однако в литературе имеются единичные описания инсулином у детей младшего возраста (до 3 лет) [5, 6].
В настоящей статье представлено описание спорадической инсулиномы у 16-летней девочки, которой была успешно выполнена лапароскопическая дистальная резекция ПЖ.
Клиническое наблюдение
Пациентка Ш., 16 лет, поступила в ФГБУ ЭНЦ в связи с жалобами на приступы потери сознания с последующей ретроградной амнезией, сопровождавшиеся слабостью и головокружением. Из анамнеза известно, что подобные приступы отмечаются в течение 3 мес. Неоднократно осматривалась врачами в момент приступа, однако гликемия не измерялась. В очередной приступ была зафиксирована потеря сознания, в связи с чем девочка была госпитализирована в реанимационное отделение, где зафиксирована гипогликемия (1,5 ммоль/л). На фоне инфузионной терапии раствором глюкозы состояние было купировано. Ребенок был переведен в эндокринологическое отделение, где при регулярном контроле гликемии зафиксированы низкие значения (2,0–2,5 ммоль/л). На магнитно-резонансной томографии (МРТ) органов брюшной полости обнаружено объемное образование в области хвоста ПЖ размерами 10 × 7 × 6 мм (рис. 1). Установлен диагноз “инсулинома пожелудочной железы? Гиперинсулинизм”. Для дальнейшего обследования ребенок был направлен в НИИ детской эндокринологии ФГБУ ЭНЦ.
На момент поступления в ФГБУ ЭНЦ при объективном осмотре патологии не выявлено. В отделении была проведена проба с голоданием, по результатам которой диагностирован органический гиперинсулинизм: через 14 ч отмечен повышенный уровень инсулина (31,6 мкЕд/мл) на фоне гипокетотической гипогликемии (гликемия – 2,4 ммоль/л, кетонемия – 0,1 ммоль/л). Мультиспиральная компьютерная томография (МСКТ) брюшной полости – в хвосте ПЖ имеется овальной формы образование размерами 12 × 10 мм, плотность образования – 175 ед.Н (рис. 2). Заключение: образование хвоста поджелудочной железы (нейроэндокринная опухоль, Grage I?).
Рис. 1. Магнитно-резонансная томограмма. В области задних отделов хвоста ПЖ определяется округлой формы слабогиперинтенсивный на Т2ВИ, слабогипоинтенсивный на Т1ВИ очаг, накапливающий контраст в артериальную фазу, быстро вымывающий его, размерами 9 × 7 × 6 мм (стрелка).
Рис. 2. Компьютерная томограмма. Структура ПЖ неоднородна за счет наличия в хвосте овальной формы гиперконтрастного в артериальную фазу образования размерами 12 × 10 мм (стрелка).
Девочке были исследованы основные биохимические и гормональные маркеры составляющих синдрома МЭН1, отклонений не выявлено. Также было проведено молекулярно-генетическое исследование гена MEN1 – мутаций в исследованном гене не выявлено. В гормональном анализе крови показатели в пределах референтных значений: паратгормон 34,58 пг/мл, ИПФР-1 542,1 нг/мл, СТГ 3,74 нг/мл, пролактин 186,1 мЕд/л, кортизол 150 нмоль/л, АКТГ 14,76 пг/мл, св.Т4 12,58 пмоль/л, ТТГ 1,034 мМЕ/л. По данным ультразвукового исследования (УЗИ) щитовидной железы и паращитовидных желез объемных образований не выявлено. На МРТ головного мозга гипофизарно-селлярная область без изменений. На период предоперационной подготовки с целью профилактики гипогликемического синдрома девочке был назначен прогликем (диазоксид) в дозе 100 мг/сут, на фоне которого отмечалась стабилизация показателей гликемии. С диагнозом “органический гиперинсулинизм. Инсулинома поджелудочной железы” больная переведена в ДГКБ № 7 им. З.А. Башляевой для проведения оперативного лечения.
13.07.2016 ребенку под интубационным наркозом выполнено оперативное вмешательство – лапароскопическая резекция ПЖ, дренирование сальниковой сумки. Под пупком введен 5-миллиметровый троакар, наложен карбоксиперитонеум под давлением 14 мм рт. ст. и потоком 5,5 л/мин, введена 5-миллиметровая оптика. Дополнительные 5-миллиметровые троакары введены справа и слева от пупка и в левой люмбодорзальной области. Желудок по большой кривизне временно фиксирован к передней брюшной стенке тракционными швами. Широко рассечена желудочно-ободочная связка. По верхнему и нижнему краю в области хвоста ПЖ рассечен задний листок брюшины. Позади железы создан туннель, через который проведен ниппельный катетер и железа отведена кпереди (рис. 3). С помощью монополярной коагуляции хвост железы отделен от селезеночной артерии и вены. Используя EnSeal, выполнена поперечная резекция на границе тела и хвоста ПЖ. Культя ПЖ ушита непрерывным обвивным швом PDS 3-0 (рис. 4). Смена 5-миллиметрового троакара в околопупочной области на 10-миллиметровый троакар, через который резецированный фрагмент ПЖ удален из брюшной полости. Сальниковая сумка дренирована трубчатым дренажом через левый троакарный доступ. Троакары удалены. Швы на раны. Асептические повязки. Время операции – 1ч 40 мин. Кровопотеря – 50,0 мл.
Рис. 3. Интраоперационное фото. ПЖ взята на держалку и отведена кпереди от селезеночных сосудов.
Рис. 4. Интраоперационное фото. Ушивание культи ПЖ обвивным швом нитью PDS II 4-0.
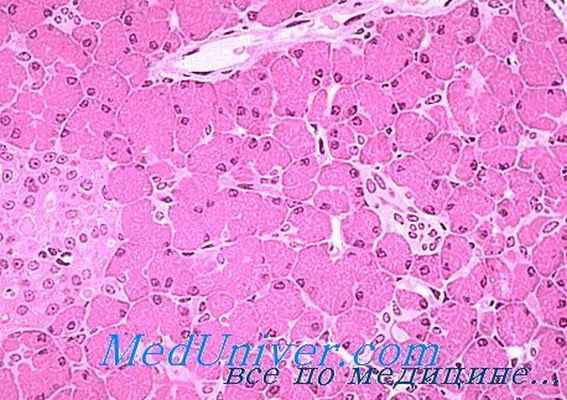
При макроскопическом исследовании в резецированном фрагменте ПЖ выявляется узел белесоватого цвета размерами 1 × 1 см с капсулой плотной консистенции. При микроскопическом исследовании определяется узел трабекулярного строения без прорастания в капсулу (рис. 5). Вне опухоли отмечается выраженная гиперплазия островков Лангерганса. При проведении иммуногистохимического исследования отмечается позитивная реакция опухолевых клеток к маркерам нейроэндокринной дифференцировки – с антителами к инсулину (рис. 6), синаптофизину и хромогранину А, индекс пролиферации Ki-67 (MIBI) – 1%. Заключение: нейроэндокринная опухоль ПЖ G1 (материал консультирован врачом отдела фундаментальной патоморфологии ФГБУ ЭНЦ Л.С. Селивановой).
Рис. 5. Поджелудочная железа. Четко отграниченная капсула от опухолевого узла. Окраска гематоксилином и эозином. ×100.
Рис. 6. Поджелудочная железа. Иммуногистохимическое исследование с антителами к инсулину, позитивная реакция. ×400.
В послеоперационном периоде отмечена эугликемия, не требующая дополнительной терапии. На 5-е сутки отмечено увеличение до 150 мл отделяемого по дренажу из сальниковой сумки с содержимым альфа-амилазы 30902 ед/л. Наряду с проводимой антибактериальной терапией к лечению добавлен октреотид (сандостатин) по 100 мкг 3 раза в день. С 12-х суток из дренажа отмечено значительное уменьшение отделяемого, которое приобрело серозный характер. Показатели амилазы в экссудате – 171 ед/л. При контрольном УЗИ органов брюшной полости свободной жидкости в проекции ПЖ не выявлено. Дренаж удален на 15-е сутки. Выписана на 20-е сутки в удовлетворительном состоянии.
Инсулиномы секретируют эндогенный инсулин автономно и независимо от уровня глюкозы в крови, что приводит к гиперинсулинемии. Клиническая картина характеризуется наличием триады Уиппла, которая включает в себя характерные симптомы гипогликемии, лабораторную гипогликемию и полную нормализацию состояния после внутривенного введения глюкозы. Симптомы гипогликемии могут быть как нейрогликопеническими (слабость, вялость, сонливость, помрачение сознания), так и адренергическими (тремор, повышенный аппетит, агрессия, беспокойство, судороги) [2, 6]. Помимо этого, в случае длительного течения заболевания у многих пациентов отмечается прогрессирующий набор избыточной массы тела на фоне гиперфагии. В отличие от взрослых пациентов клинически заподозрить гипогликемию у детей бывает сложно, что обусловлено неспособностью детей детально описать свои жалобы. Неспецифичность симптоматики гипогликемии, высокие энергозатраты в детском возрасте, а также отсутствие инсулинорезистентности обусловливают высокую частоту судорожного синдрома у детей на момент первичной диагностики.
Критерием постановки диагноза является наличие лабораторно подтвержденного органического гиперинсулинизма (инсулин плазмы в момент гипогликемии >2,0 Ед/л) при выявлении новообразования ПЖ [1, 4]. Дифференциальная диагностика инсулином у детей должна проводиться с ятрогенными гипогликемиями (подколки инсулина, прием сахароснижающих препаратов) и врожденным гиперинсулинизмом. Для последнего характерен ранний возраст манифестации (до 3 лет).
Инсулиномы ассоциированы с синдромом МЭН1 в 5–10% случаев у взрослых и в 30–50% случаев у детей, однако они редко являются его первым проявлением [1, 3]. Синдром МЭН1 – это наследственное заболевание, вызванное аутосомно-доминантными мутациями в гене-супрессоре опухолевого роста MEN1. В структуру синдрома МЭН1 входят опухоли паращитовидных желез (они выявляются в более чем 90% всех случаев), различные гастроэнтеропанкреатические нейроэндокринные опухоли (выявляются у 50–60% больных), опухоли аденогипофиза (50%) и надпочечников (40%). Заболевание встречается с частотой 1:30000 – 1:40000, чаще всего проявляется в возрасте 20–30 лет, а первым его симптомом является гиперпаратиреоз [3]. Инсулиномы, ассоциированные с синдромом МЭН1, как правило, множественные, мелкие (до 1 см) и доброкачественные, они могут сочетаться с гормонально неактивными опухолями ПЖ. Верификация диагноза МЭН1 во многом определяет тактику ведения пациента: объем хирургического вмешательства, скрининг на другие составляющие синдрома, а также генетическое консультирование родственников [3, 5]. Учитывая высокий риск наличия синдромального варианта, всем детям с инсулиномами показано молекулярно-генетическое исследование гена MEN1.
Ангиографические исследования, включая Са2+ССА, до недавнего времени активно использовались в топической диагностике инсулином, однако в силу инвазивности этих методов и высокого риска развития осложнений в настоящее время к ним прибегают лишь в исключительных случаях. В последние годы в практику активно внедряются изотопные исследования, из которых в диагностике инсулином информативными считаются сцинтиграфия рецепторов соматостатина, а также ПЭТ с 18F-ДОПА [6].
С точки зрения морфологии инсулиномы – это типичные нейроэндокринные опухоли. При иммуногистохимических исследованиях инсулиномы позитивно окрашиваются на инсулин, проинсулин, хромогранин А, синаптофизин, нейронспецифическую энолазу, цитокератин и Ki-67 (Mib-1) [1]. Определенные гистохимические маркеры или гистохимические параметры, отражающие биологическое поведение опухоли, отсутствуют, и в связи с этим определение злокачественности инсулиномы бывает основано на выявлении отдаленных метастазов и появлении признаков локальной инвазии [4].
Основным методом лечения инсулином является хирургический, с успехом применяемый у 77–100% больных [2, 10]. Медикаментозное лечение в основном применяется для контроля уровня сахара крови в предоперационном периоде, у тех, кому противопоказана операция, или больных с нерезектабельными метастазами в печень [2, 4]. Диазоксид (50–600 мг/день) является наиболее эффективным препаратом для контроля гипогликемии (50–60% симптомов), и его действие основано на прямой супрессии продукции инсулина бета-клетками ПЖ.
Учитывая тот факт, что 90% инсулином являются доброкачественными солитарными опухолями размерами менее 2 см, энуклеация опухоли, когда это возможно, может считаться оптимальным объемом хирургического вмешательства [2, 11]. По данным литературы, энуклеация инсулиномы бывает возможна в 34–54% случаев [6]. В то же время резекция ПЖ (тотальная или частичная) может быть показана при инвазии в окружающие анатомические структуры и главный проток ПЖ, сомнении в доброкачественном характере поражения, а также при вовлечении в патологический процесс региональных лимфоузлов [2]. При выполнении дистальной резекции ПЖ следует стремиться сохранять селезенку, особенно у детей, что приводит к уменьшению числа послеоперационных инфекционных осложнений [12]. При локализации опухоли в головке ПЖ у 5,5–16% больных может потребоваться панкреатодуоденальная резекция [2, 6]. Альтернативой этому у детей может быть резекция головки ПЖ с сохранением двенадцатиперстной кишки и наложением дистального панкреатоеюноанастомоза, что было использовано у больных с фокальной формой врожденного гиперинсулинизма [13].
В представленном клиническом наблюдении был выбран вариант дистальной резекции ПЖ, исходя из установленной локализации опухоли вблизи главного протока ПЖ. Подобный объем вмешательства, на наш взгляд, в большей степени гарантирует от возникновения стойкого наружного панкреатического свища.
При хирургическом лечении в большинстве наблюдений используются традиционные открытые лапаротомные вмешательства, преимуществом которых считают возможность провести интраоперационную пальпацию всей ПЖ для выявления опухоли, особенно при множественных локализациях инсулином [2, 6]. В последние годы стали появляться публикации об успешном использовании у взрослых больных с инсулиномами лапароскопическогодоступа с интраоперационным УЗИ ПЖ [11, 14]. В доступной нам литературе мы нашли только два наблюдения применения лапароскопических технологий у детей с инсулиномами: в одном случае – энуклеации опухоли, в другом – дистальной резекции ПЖ [5, 15].
Одним из специфических осложнений хирургических вмешательств на ПЖ является возникновение наружного панкреатического свища или перипанкреатических скоплений [6, 16]. Частота развития подобных осложнений достигает 23,7%, при этом значительных различий как между энуклеацией и дистальной резекцией, так и между открытыми и лапароскопическими операциями не отмечено [2, 6]. Для профилактики свищей после энуклеации инсулиномы предлагают использовать различные фибринные клеи или накладывают на паренхиму ПЖ матрасные швы. При выполнении лапароскопических резекций ПЖ оптимальным является использование сшивающих аппаратов, что было применено у детей с травматическими разрывами ПЖ [17]. По большей части панкреатические свищи могут закрываться спонтанно, что и отмечено в представленном нами наблюдении.
Злокачественные формы инсулином встречаются в 10% случаев [4, 18]. Наиболее часто метастазы при этом выявляются в печени, регионарных лимфоузлах, реже – в костях и брюшине. Хирургическое иссечение метастазов может продлевать выживаемость больных со злокачественными инсулиномами [2, 4].
Пациенты с доброкачественной инсулиномой имеют благоприятный прогноз с успехом хирургического лечения в 77–100% случаев [10]. В противоположность этому у пациентов со злокачественными инсулиномами, несмотря на все современные технологии, средняя выживаемость составляет около 2 лет [2, 4].
Заключение
Представленное клиническое наблюдение свидетельствует о необходимости настороженности в отношении инсулиномы у детей и подростков с приступами гипогликемии. Использование инструментальных методов, таких как МСКТ и МРТ, позволяет поставить правильный топический диагноз. Лапароскопическая резекция поджелудочной железы у детей с инсулиномой является малотравматичным и эффективным оперативным вмешательством.
Дополнительная информация
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи. Медицинские данные опубликованы с письменного согласия пациента.
Морфология гиперинсулинизма. Патологическая анатомия инсуломы
Морфология гиперинсулинизма. Патологическая анатомия инсуломы
Это патологическое состояние характеризуется абсолютным или относительным избытком инсулина в крови, клинически проявляющимся гипогликемией различной степени выраженности. Функциональные формы гиперинсулинизма с явлениями вторичной гипогликемии, которые вызываются внепанкреатическими факторами, обычно не вызывают заметных патоморфологических изменений в островковых клетках. В основе истинного гиперин-сулипизма лежит гормональноактивная опухоль, развивающаяся из клеток островков Лангерганса поджелудочной железы, получившая название инсуломы или незидиобластомы. Эта опухоль, в избытке продуцирующая инсулин, была описана в 1901 г. Л. В. Соболевым, а в 1902 г.— А. Николзом.
Возникнуть инсуломы могут из эмбриональных зачатков, из которых образуется островковый аппарат поджелудочной железы, а также из эктопической инсулярной паренхимы различной степени зрелости и дифференцировки. Предполагают, что эти гормональноактивные опухоли развиваются из бета-клеток островков Лангерганса, в отличие от функционально неактивных новообразований поджелудочной железы, развивающихся из альфа-клеток, вследствие чего может возникнуть сахарный диабет (Н. Ferner, В. С. Жданов, А. И. Абрикосов).
В преобладающем числе случаев (примерно в 90%) эти гормональноактивпые опухоли имеют доброкачественный, аденоматозный характер. Гиперинсулинизм чаще вызывается одиночной опухолью; множественные новообразования из островковой паренхимы встречаются значительно реже; в порядке же исключения описываются опухоли, возникающие из островковых клеток добавочной поджелудочной железы (I. Howard, N. Moss, T. Rhoads, В. В. Виноградов).
При макроскопическом исследовании инсуломы чаще имеют вид небольшого солитарного узла. Опыт применения серийных срезов для тщательного изучения поджелудочной железы показал, что эти гормональноактивные опухоли встречаются значительно чаще. Это новообразования обычно овальной или округлой формы, плотной консистенции, бурого или серо-красного цвета (чаще обусловленного диффузными кровоизлияниями). Локализуются они преимущественно в хвостовой части и теле поджелудочной железы и, как правило, заключены в соединительнотканную капсулу. Иногда поражается и головка железы. Инсуломы чаще всего имеют альвеолярное или солидное строение, однако встречаются гормональноактивные опухоли поджелудочной железы с папилломатозным и тубулярным строением.
Особенностью этих опухолей является разнообразие клеточного состава. Наиболее часто опухолевые клетки в инсуломе небольших размеров, цилиндрической или полигональной формы с очень бледной протоплазмой. Ядра клеток округлой формы, сочные, иногда гиперхромные, фигуры митоза, как правило, не обнаруживаются. Считается, что доброкачественные инсуломы состоят из островковых клеток, очень близких к нормальным, с высокой степенью дифференцировки. Поэтому иногда эта опухоль по своему строению до некоторой степени напоминает гигантские островки Лангерганса.
В интерстициальной ткани этой опухоли могут обнаруживаться признаки гиалиноза, кровоизлияний, а также различные внеклеточные дистрофические изменения с резким развитием фиброзных элементов (A. Whipple, Б. С. Розанов, З. А. Топчиа-швили). О. В. Николаев и Э. Г. Вейнберг на основании анализа гистологической структуры инсулом выделяют 3 основных типа опухоли: 1) паренхиматозный, 2) фиброзный, 3) смешанный.
Первый тип характеризуется наличием сплошной массы ост-ровковой ткани. Эпителиальные клетки островков образуют розетки, нередко располагаются вокруг сосудов, а в протоплазме клеток выявляется зернистость, характерная для бета-клеток. Иногда наблюдается почти полная дегрануляция таких клеток, что, очевидно, связано с выбросом инсулина в кровь.
Опухоли паренхиматозного типа обильно васкуляризированы, в различных участках встречаются кровоизлияния и некротический распад. Комплексы железистоподобных структур составляют основную массу опухолей, однако бывают и пучки гиалинизированных соединительнотканных волокон. Инсуломы фиброзного типа характеризуются мощным развитием соединительной ткани, среди которой обнаруживаются опухолевые клетки, образующие тяжи. Часть клеток содержит гранулы, большинство из них дегранулировано. Для опухолей фиброзного типа свойственно гиалиновое перерождение волокон и стенок кровеносных сосудов.
Инсуломы смешанного типа построены из элементов, характерных как для паренхиматозной, так и фиброзной формы.
Для ультраструктуры бета-клеток, образующих аденому, характерно чрезвычайно малое содержание органелл и гранул. Вокруг ядра этой клетки определяются своеобразные нитевидные образования. В клетках доброкачественных инсулом поджелудочной железы обнаруживаются довольно крупные поля Гольджи. В цитоплазме опухолевых клеток передко, как отмечают М. Greider, В. Zollinger, D. Elliot, E. Passaro, S. Lazarus, В. Volk, M. Greider и сотр., встречаются кристаллоподобные структуры, локализующиеся в области полюса, обращенного к капилляру, в то время как на противоположном полюсе обычно располагается эндоплазматический ретикулум.
Согласно наблюдениям В. Honjin, A. Takahashi, H. Murajama, S. Murono, Т. Hanyn, A. Like, I. Steinke, E. Jones, G. Cahill, гранулы бета-клеток сравнительно крупные и часто имеют форму мешочка с разрыхленным содержимым. Ядра аденоматозных клеток округлой формы, иногда несколько овальные или даже бобовидные, содержат диффузно распределяющуюся зернистость; в некоторых ядрах выявляются фибриллярные структуры. При электронномикроскопическом исследовании S. Bencos-me, В. Allen, H. Latta, W. Gusek, P. Lacy, S. Williamson, P. Lacy отмечали, что бета-клетки снаружи окружены зазубренными мембранами, создающими тесный контакт между клеточными образованиями.
В инсуломах поджелудочной железы альфа-клетки (I. Iniery, I. Bader, Е. Porta, К. Yerry, В. Scott, М. McGavran, В. Unger, L. Becant, F. Potet, E. Martin, J. Thiery, J. Bader, S. Bonfils, A. Lambling) отличаются наличием и цитоплазме крупных электронпоплотных гранул, а также увеличением количества митохондрий. Эндоплазматическая сеть в этих клетках неравномерно уплотнена, ядро и ядрышки не изменены.
Морфология гиперинсулинизма. Патологическая анатомия инсуломы
НИИ детской эндокринологии Эндокринологического научного центра, Москва
Журнал: Проблемы эндокринологии. 2010;56(6): 41‑47
Меликян М.А. Врожденный гиперинсулинизм. Проблемы эндокринологии. 2010;56(6):41‑47.
Melikian MA. Congenital hyperinsulinism. Problemy Endokrinologii. 2010;56(6):41‑47. (In Russ.).
НИИ детской эндокринологии Эндокринологического научного центра, Москва
Врожденный гиперинсулинизм (ВГИ) - одна из основных причин развития персистирующих гипогликемических состояний в детском возрасте. Биохимически ВГИ характеризуется неадекватной гиперсекрецией инсулина β-клетками поджелудочной железы. ВГИ является гетерогенным заболеванием в отношении как клинических проявлений и морфологических форм, так и молекулярно-генетических дефектов, лежащих в его основе. В настоящей статье изложены современные взгляды на основные механизмы развития ВГИ, представлена клиническая характеристика заболевания, предложены международные протоколы обследования и лечения детей, страдающих данной патологией.
НИИ детской эндокринологии Эндокринологического научного центра, Москва
Врожденный гиперинсулинизм (ВГИ) — наследственное заболевание, характеризующееся неадекватной гиперсекрецией инсулина β-клетками поджелудочной железы, что приводит к развитию персистирующих гипогликемических состояний. В литературе описаны 8 генов, участвующих в развитии ВГИ. От 40 до 60% случаев ВГИ связаны с дефектами генов KCNJ11 и ABCC8, кодирующих белки, которые участвуют в работе АТФ-зависимых калиевых каналов β-клеток поджелудочной железы. Около 15—20% связаны с активирующими мутациями в генах GCK и GLUD1, участвующих в регуляции внутриклеточного метаболизма глюкозы. В литературе [1] также имеются единичные описания случаев ВГИ, связанных с дефектами генов HADH, HNF4α, INSR, UCP2. В 30—40% всех случаев ВГИ не удается выявить молекулярно-генетических дефектов в указанных генах.
Распространенность ВГИ варьирует от 1:30 000 до 1:50 000 новорожденных, а в популяциях с высоким уровнем близкородственных браков достигает 1:2500 новорожденных [2, 3].
ВГИ был впервые описан как «идиопатическая гипогликемия детского возраста» ученым I. MacQuarrie [4] в 1954 г. В дальнейшем ВГИ обозначали такими терминами, как, например, «лейцин-чувствительная гипогликемия», «синдром дисрегуляции β-клеток», «персистирующие гиперинсулинемические гипогликемии младенческого возраста». Длительное время для определения ВГИ использовался термин «незидиобластоз» [3]. Этот термин был введен Г. Лейдло еще в 1938 г.
Незидиобластоз — тотальная трансформация протокового эпителия поджелудочной железы в β-клетки, продуцирующие инсулин. К настоящему времени доказано, что подобная морфологическая картина является нормальной в младенческом возрасте и не служит причиной гиперинсулинизма [5].
Морфологически ВГИ разделяют на 3 основные формы: диффузную, при которой поражены все β-клетки поджелудочной железы, фокальную, если очаг поражения ограничен небольшим участком гиперплазированных клеток, содержащих крупные ядра, и атипичную [6, 7].
Истинной причиной гиперсекреции инсулина при ВГИ является чаще всего неадекватная работа АТФ-зависимых К-каналов β-клеток поджелудочной железы, что обусловлено молекулярно-генетическими дефектами генов KCNJ11 и ABCC8 [1].
Нарушения функции АТФ-зависимых К-каналов, а также дефекты регуляции внутриклеточного метаболизма глюкозы могут приводить к развитию гиперинсулинемических гипогликемических состояний. Наиболее частой причиной ВГИ являются инактивирующие мутации генов KCNJ11 и ABCC8 [8—11].
АТФ-зависимые калиевые каналы β-клеток представляют собой октамерные структуры, внутренние отделы которых представлены 4 субъединицами белка Kir6.2, кодируемого геном KCNJ11, а наружные — 4 субъединицами белка SUR1, кодируемого геном ABCC8. Данные каналы способны изменять степень поляризации мембраны клетки. Функциональная активность каналов регулируется уровнем внутриклеточных адениновых нуклеотидов. Инактивирующие мутации генов KCNJ11 и ABCC8 приводят к закрытию данных каналов, что влечет за собой избыточное поступление Cа 2+ в клетку и гиперсекрецию инсулина [1, 8].
Описаны как аутосомно-рецессивные, так и аутосомно-доминантные мутации указанных генов. К настоящему моменту выявлено более 150 мутаций в гене ABCC8 и 25 мутаций в гене KCNJ11 [12].
ВГИ, связанный с рецессивными мутациями генов KCNJ11 и ABCC8, характеризуется тяжелым течением, ранним дебютом гипогликемии и, как правило, не поддается консервативной терапии. Доминантно наследуемые формы протекают мягче, манифестируют позже и в большинстве случаев чувствительны к терапии диазоксидом [1, 6, 13].
Помимо нарушений работы АТФ-зависимых калиевых каналов β-клеток причинами развития ВГИ могут служить нарушения работы ферментов, участвующих во внутриклеточном метаболизме глюкозы. К ним относятся глюкокиназа, глутаматдегидрогеназа и 3-гидрокси-ацилКоА-дегидрогеназа.
Глюкокиназа — один из важных регуляторных факторов секреции инсулина. Данный фермент катализирует реакцию фосфорилирования глюкозы в ее активный метаболит — глюкозо-6-фосфат. Активирующие доминантные мутации гена GCK приводят к увеличению экспрессии фермента, что влечет за собой гиперсекрецию инсулина [14]. Данная форма ВГИ характеризуется вариабельностью клинической картины. Описано бессимптомное течение. Некоторые мутации проявляют себя лишь гипогликемическими состояниями после приема пищи при сохранении нормального уровня глюкозы крови натощак. Существуют также описания тяжелых, резистентных к терапии, форм [15].
Митохондриальный фермент глутаматдегидрогеназа (кодируется геном GLUD1) катализирует реакцию превращения глутамина в α-кетоглутарат и аммоний. Мутации гена GLUD1 ослабляют чувствительность фермента к лейцину, который является его специфичным ингибитором, что приводит к повышению активности фермента и избыточной продукции АТФ за счет активирующего влияния лейцина и аммония на реакции цикла Кребса [16]. Отмечается повышение уровня аммиака крови. Эта форма ВГИ также носит название гипераммониемийной лейцинчувствительной гипогликемии. Мутации в гене GLUD1 наследуются по аутосомно-доминантному типу. Гипогликемические состояния при дефектах глутаматдегидрогеназы купируются низкопротеиновой диетой, хорошо поддаются терапии диазоксидом [17, 18].
Другой редкой причиной рецессивно наследуемого ВГИ являются дефекты гена HADH, кодирующего фермент 3-гидрокси-ацилКоА-дегидрогеназу. Данный фермент катализирует предпоследнюю реакцию в процессе β-окисления короткоцепочечных жирных кислот, в результате которой образуется 3-кето-ацилКоА. Инактивирующие мутации гена HADH приводят к гиперпродукции инсулина и избыточному накоплению продуктов кетогенеза. Механизм гиперинсулинизма при этих мутациях остается неясным. Это единственная форма гиперинсулинемической гипогликемии, протекающая с кетозом. Характерно повышение уровня 3-гидроксибутирил-карнитина в крови и 3-гидроксиглутарата в моче. Как правило, течение мягкое и отмечается хороший терапевтический эффект от диазоксида [19, 20].

Фокальные формы ВГИ формируются в случае соматического снижения гомозиготности унаследованной от отца мутации в генах ABCC8 и KCNJ11 и специфической потери материнской аллели в регионе импринтинга на 11р 15. При этом происходит изменение экспрессии импринтинговых генов в регионе 11р 15.5: снижается экспрессия генов H19 и Р57KIP2, являющихся супрессорами опухолевого роста, и увеличивается экспрессия гена, кодирующего инсулиноподобный фактор роста 2-го типа (IGF2), являющийся мощным фактором пролиферации клеток. Подобное сочетание нарушений импринтинга с наследованием мутации в генах KCNJ11 или ABCC8 приводят к развитию фокального аденоматоза ткани поджелудочной железы [21]. Данные формы заболевания составляют около 40% всех случаев ВГИ [1]. По своим клиническим проявлениям фокальный ВГИ не отличается от диффузного. При своевременной постановке молекулярно-генетического диагноза и визуализации образования возможно хирургическое лечение в виде селективной резекции фокуса, что приводит к полному выздоровлению [21, 22]. Основные генетические формы ВГИ представлены в табл. 1.
Клиническая картина. ВГИ, как правило, манифестирует в неонатальный период, однако возможен и более поздний дебют, вплоть до 3-летнего возраста. Чем раньше проявляется заболевание, тем тяжелее оно протекает [6, 23]. Гипогликемические состояния при ВГИ обычно носят тяжелый характер и быстро приводят к развитию судорог и потере сознания. Описаны и мягкие формы, протекающие почти бессимптомно, проявляющиеся лишь гиподинамией и сниженным аппетитом. В связи с избыточной продукцией инсулина еще во внутриутробном периоде дети с ВГИ, как правило, рождаются крупными. При рождении часто выявляется макросомия, кардиомиопатия, гепатомегалия [7]. У матерей может отмечаться избыточная прибавка массы тела во время беременности. Для поддержания нормогликемии детям с ВГИ требуются крайне высокие дозы глюкозы. Потребность во внутривенной инфузии раствора глюкозы может достигать 20 мг/кг/мин [6, 7, 23].

Дифференциальная диагностика. ВГИ необходимо дифференцировать от других форм гипогликемии, таких как врожденные дефекты β-окисления жирных кислот; синдромальные формы гиперинсулинизма (синдром Беквита—Видемана, синдром Сотоса, синдром Ашера и др.), от врожденных заболеваний гликозилирования и инсулинпродуцирующими опухолями поджелудочной железы, дефицита контринсулярных гормонов, гликогеновых болезней печени, дефектов кетогенеза и глюконеогенеза. Помимо этого, не стоит забывать про транзиторные формы неонатального гиперинсулинизма, связанные с диабетической фетопатией, задержкой внутриутробного развития и перинатальной асфиксией [24]. Схемы дифференциальной диагностики основных форм гипогликемического синдрома представлены в табл. 2.

Лечение. Основной целью лечения ВГИ является поддержание стойкой нормогликемии (3,5—6,0 ммоль/л). Даже единичные эпизоды гипогликемических состояний в первые месяцы жизни могут быть чреваты тяжелыми неврологическими осложнениями. В связи с высокой потребностью в глюкозе пациентам с ВГИ рекомендована постановка центрального катетера, дающая возможность введения больших объемов концентрированного раствора. Рекомендовано дробное кормление обогащенными углеводами продуктами. Некоторым пациентам требуется постановка желудочного зонда для адекватного питания [24, 27, 28]. Блок кетогенеза, вызванный гиперсекрецией инсулина, лишает детей с ВГИ альтернативных ресурсов энергии для головного мозга, что крайне быстро приводит к развитию судорог и в отсутствие адекватного лечения к формированию автономной эпилепсии. По последним международным рекомендациям [27], пациентам с ВГИ показано поддерживать уровень глюкозы в крови не ниже 3,8—4,0 ммоль/л. Среди лекарственных препаратов, применяемых для лечения гиперинсулинемических гипогликемических состояний, препаратом выбора является диазоксид [28]. Диазоксид — агонист АТФ-зависимых К-каналов β-клеток поджелудочной железы. Механизм его действия заключается в активации работы самих каналов. Эффективность терапии варьирует в зависимости от молекулярно-генетических дефектов. Большинство пациентов с рецессивно наследуемыми мутациями генов KCNJ11 и ABCC8, а также некоторыми мутациями гена GCK, резистентны к данному лечению [6, 13, 28]. Для потенцирования действия диазоксида в некоторых случаях возможно присоединение хлортиазида. Нифедипин, являясь блокатором кальциевых каналов, имеет супрессорный эффект на секрецию инсулина. Его эффективность в лечении ВГИ крайне низкая, существуют лишь единичные описания его успешной монотерапии [7]. Соматостатин, являясь аналогом одноименного гормона, активирует специфичные рецепторы, расположенные в ткани поджелудочной железы, что подавляет секрецию инсулина. Данный препарат эффективен в сочетании с дробным режимом кормления. В редких случаях возможно использование пролонгированных форм, когда инъекция выполняется 1 раз в месяц [28, 29]. Глюкагон применяется в острых ситуациях для купирования гипогликемии. Его длительное использование возможно лишь в виде постоянной подкожной инфузии. Высокие дозы глюкагона (>20 мкг/кг/ч) вызывают обратную реакцию — выброс инсулина [30]. В табл. 3 приведены основные лекарственные препараты для лечения ВГИ.
Отдаленные наблюдения. У большинства пациентов с ВГИ, тяжесть течения и частота эпизодов гипогликемии резко снижаются с увеличением возраста. Описано множество случаев спонтанного выздоровления. В случае консервативного лечения в среднем к 3—4 годам жизни среднесуточная доза диазоксида снижается до минимальной терапевтической (5 мг/кг/сут). В литературе имеются данные, свидетельствующие о развитии с возрастом сахарного диабета у неоперированных пациентов с ВГИ [33]. Среди детей, перенесших субтотальную панкреатэктомию, около 40% имеют ИЗСД, до 60% не нуждаются в инсулинотерапии, 2—5% требуют лечения диазоксидом для поддержания нормогликемии [34]. По данным разных авторов [35], задержка психомоторного развития отмечается у 30—40% всех пациентов с ВГИ. В 15—20% случаев выявлено формирование автономной эпилепсии, при которой требуется терапия противосудорожными препаратами. Степень тяжести неврологических осложнений напрямую зависит от возраста манифестации заболевания, а также от своевременности и адекватности проводимой терапии.
В данном обзоре литературы были рассмотрены основные молекулярно-генетические предпосылки развития ВГИ, представлены современные взгляды на наличие взаимосвязей генотипа и фенотипа. На основании международного опыта предложены современные протоколы диагностики, лечения и наблюдения за детьми, страдающими ВГИ. Своевременная постановка диагноза, выбор адекватного лечения и динамический контроль позволяют минимизировать неврологические осложнения гипогликемических состояний. Несмотря на прорыв в понимании этиологии и патогенеза ВГИ, в 50% случаев молекулярно-генетический диагноз остается неясным, что требует дальнейших исследований в этой области.
Морфологические факторы прогноза нейроэндокринных опухолей желудочно-кишечного тракта и поджелудочной железы
Нейроэндокринные опухоли (НЭО) могут развиваться в любых органах, где в норме имеются эндокринные клетки: в желудочно-кишечном тракте (ЖКТ), легких, тимусе, почках, яичниках, простате, молочной и щитовидной железах, коже (1). По своему происхождению нейроэндокринные (эндокринные) опухоли пищеварительного тракта можно разделить на опухоли желудочно-кишечного тракта
(НЭО ЖКТ) и опухоли поджелудочной железы (НЭО ПЖ)
- КЛЮЧЕВЫЕ СЛОВА: опухоль, аппендикс, синдром, кишки, тимус, желудок, операция
Нейроэндокринные опухоли (НЭО) могут развиваться в любых органах, где в норме имеются эндокринные клетки: в желудочно-кишечном тракте (ЖКТ), легких, тимусе, почках, яичниках, простате, молочной и щитовидной железах, коже (1). По своему происхождению нейроэндокринные (эндокринные) опухоли пищеварительного тракта можно разделить на опухоли желудочно-кишечного тракта
(НЭО ЖКТ) и опухоли поджелудочной железы (НЭО ПЖ)
Рис. 1. А. Низкая пролиферативная активность (Ki67 = 8%) в высокодифференцированной нейроэндокринной карциноме желудка. Б. Высокая пролиферативная активность (Ki67 > 50%) в низкодифференцированной нейроэндокринной карциноме желудка
Рис. 2. А. Злокачественная инсулинома поджелудочной железы. Инвазия сосуда капсулы. Б. Злокачественная гастринома. Инвазия нерва комплексами опухолевых клеток.
Рис. 3. Ко-экспрессия цитокератина 19 (А) и синаптофизина в смешанной экзо-эндокринной карциноме поджелудочной железы. Появление метастазов в печени через 4 года после операции
Рис. 4. А. Метастаз в лимфатический узел злокачественного карциноида подвздошной кишки: многочисленные комплексы опухолевых клеток. Реакция с серотонином. Б. То же наблюдение. Низкая пролиферативная активность клеток злокачественного карциноида (Ki67 мене
НЭО соответственно своему эмбриональному происхождению делятся на опухоли верхней (карциноиды легких, тимуса, желудка и двенадцатиперстной кишки), средней (аппендикса, подвздошной, тощей и проксимальной кишки) и нижней (дистальной части толстой и прямой кишки) кишки. Эффект применения химиотерапии (ХТ) в случае нерезектабельных и метастазирующих НЭО зависит как от их функциональной активности (клинического синдрома), так и от степени злокачественности опухоли или ее биологического потенциала. Важнейшими критериями злокачественного потенциала НЭО, определяющими степень их злокачественности (Grade 1 ), считают степень дифференцировки опухоли (высокодифференцированные, низкодифференцированные), наличие инвазии сосудов и нервов, некрозов, высокую митотическую и пролиферативную активность клеток опухоли.
Степень дифференцировки и степень злокачественности (Grade) опухоли
В 2000 году Всемирной организацией здравоохранения (ВОЗ) [2] была принята классификация НЭО ЖКТ и ПЖ, где эти опухоли разделены на 3 основные группы: 1) высокодифференцированные опухоли, доброкачественные и неопределенной степени злокачественности (функционирующие и нефункционирующие); 2) высокодифференцированные эндокринные карциномы низкой степени злокачественности (функционирующие и нефункционирующие); 3) низкодифференцированные эндокринные карциномы высокой степени злокачественности (мелко- и крупноклеточные эндокринные карциномы); 4) смешанные экзо-эндокринные карциномы. В настоящее время Европейской ассоциацией по нейроэндокринным опухолям (ENETS) предложено делить НЭО ЖКТ и ПЖ по степени их злокачественности (Grade) на 3 основные группы – G1, G2, G3 (табл. 1). В соответствии с этим в группы G1-G2 входят высокодифференцированные НЭО ЖКТ и ПЖ, а в группу G3 – низкодифференцированные нейроэндокринные карциномы (НЭК) [3]. Это деление базируется на параметрах, которые непосредственно отражают степень злокачественности НЭО – индексах митотической и пролиферативной активности опухолевых клеток. Индекс митотической активности может быть подсчитан при рутинном исследовании в препаратах, окрашенных гематоксилином и эозином, он определяется как количество митозов в 10 репрезентативных полях зрения (РПЗ) с равномерным распределением в них опухолевых клеток, без артефициальных изменений и при большом увеличении микроскопа (х400). Индекс пролиферации определяется при иммуногистохимическом (ИГХ) исследовании с использованием антител Ki67 (клон MIB-1) как доля клеток, ядра которых экспрессируют. Этот маркер из расчета на 100 опухолевых клеток (%) при большом увеличении микроскопа (х400). Индекс Ki67 вычисляется как среднее значение при просчете не менее 1000 клеток (оптимально- 2000 клеток). Считается, что митотический индекс более точно отражает пропорцию делящихся клеток и, следовательно, является более надежным критерием степени злокачественности НЭО, чем индекс Ki67 (4). Поэтому в морфологическом заключении при исследовании НЭО обязательно следует указывать значения и митотического индекса и индекса пролиферации. Чтобы выделить опухоли, для лечения которых рекомендовано применение более агрессивных методов терапии, следует проводить учет пролиферирующих клеток не в одном произвольно выбранном участке опухоли, а обязательно исследовать наиболее активно пролиферирующие участки опухоли – так называемые «горячие точки».
Высокодифференцированные НЭО ЖКТ и ПЖ – это опухоли низкой или промежуточной степени злокачественности, а низкодифференцированные – высокой. Низкодифференцированные НЭО имеют тенденцию к стремительной диссеминации, они устойчивы к терапии, быстро приводят к летальному исходу. Поэтому самым важным аспектом морфологического заключения является принципиальное разграничение высокодифференцированных и низкодифференцированных НЭО (рис. 1, А и Б). Высокодифференцированные НЭО – доброкачественные и неопределенной степени злокачественности (пограничные) – характеризуются слабо выраженной клеточной атипией; в высокодифференцированных эндокринных карциномах низкой степени злокачественности клеточная атипия слабо или умеренно выражена, ядра гиперхромные с отчетливыми ядрышками, характерно увеличение ядерно-цитоплазматического соотношения, митотической активности и индекса пролиферации Ki67. Большинство высокодифференцированных НЭО ЖКТ и ПЖ прогрессируют медленно, иногда в течение многих лет и даже десятилетий, но, тем не менее, являются потенциально злокачественными новообразованиями. В последние годы накоплены данные о том, что метастазы, в том числе и в печень, могут выявляться через много лет (10-30 и более) после удаления так называемых «доброкачественных» НЭО 9. Поэтому сейчас рекомендовано вообще отказаться от термина «доброкачественные НЭО» и использовать термин «НЭО неопределенной степени злокачественности» [4].
Функционирующие и нефункционирующие НЭО
Для определения функционального статуса НЭО при гистологическом исследовании используют эндокринные маркеры – пептиды и/или амины, обладающие гормональной активностью (инсулин, глюкагон, соматостатин, вазоактивный интестинальный полипептид, панкреатический полипептид, гастрин, серотонин, АКТГ, кальцитонин, и другие) [10]. НЭО часто продуцируют сразу несколько гормонов, поэтому диагноз базируется как на наличии характерного гиперфункционального синдрома, так и на выявлении доминирующей популяции эндокринных клеток, составляющей более 50% клеток опухоли. В соответствии с этим НЭО верифицируют как инсулиномы, глюкагономы, гастриномы, соматостатиномы, ПИПомы, ВИПомы, кальцитониномы, карциноиды (серотонин-продуцирующие) и др. Важная информация о функциональном статусе опухоли может быть получена при исследовании ультраструктуры опухолевых клеток. Тип эндокринных гранул в их цитоплазме и их количество позволяют уточнить диагноз НЭО, особенно при нефункционирующих опухолях.
Для НЭО различной локализации имеются и свои специфические факторы прогноза, которые следует учитывать при постановке диагноза и выборе терапии.
Нейроэндокринные опухоли ПЖ (НЭО ПЖ)
Большинство высокодифференцированных функционирующих НЭО пищеварительного тракта составляют опухоли ПЖ (табл. 2). Опухоли, которые преимущественно локализуются в теле и хвосте ПЖ (глюкагономы, инсулиномы, ВИПомы), имеют склонность к гематогенной диссеминации; гастриномы ПЖ чаще метастазируют в регионарные лимфатические узлы. НЭО ПЖ, продуцирующие островковые гормоны (инсулин, глюкагон, соматостатин и панкреатический полипептид), как правило, имеют менее злокачественный потенциал, чем те, которые продуцируют эктопические гормоны, в норме не характерные для этого органа (гастрин, нейротензин, АКТГ, кальцитонин, гормон роста и другие).
Гастриномы ПЖ обладают более злокачественным потенциалом, чем соответствующие опухоли 12-перстной кишки. Хотя обычно они прогрессируют медленно, но в большинстве случаев дают метастазы в лимфатические узлы или печень. Иммунофенотип клеток гастрином ПЖ характеризуется ко-экспрессией маркеров нейроэндокринной и экзокринной дифференцировки (хромогранина А, синаптофизина, цитокератинов 19 и 20 и/или ЭМА), следовательно, они обладают смешанным экзо-эндокринным иммунофенотипом, который и определяет их злокачественный потенциал [11, 12]. В последние десятилетия применение эффективных противоязвенных препаратов в лечении пациентов с гастриномами позволяет смягчить симптомы эндокринной гиперфункции, однако это не устраняет саму причину опухоли [13]. Это привело к тому, что во всех индустриально развитых странах в последние годы возросла летальность от гастрином, обусловленная их латентным ростом и прогрессированием. Риск смерти у пациентов с гастриномами ПЖ увеличивается при высоком уровне гастрина в сыворотке, наличии метастазов в лимфатические узлы, печень, кости, при крупных размерах опухоли, а также при ее поздней диагностике.
Глюкагономы ПЖ долго могут расти как нефункционирующие опухоли, а симптомы синдрома Маллисона (некротическая мигрирующая эритема, глоссит, хейлит, анемия, снижение веса, депрессия и венозный тромбоз) возникают, как правило, в тех случаях, когда опухоль достигает достаточно больших размеров. В этой стадии уже имеются метастазы и плохой прогноз заболевания. Соматостатиномы ПЖ также могут долго не давать симптомов. Характерный для этой опухоли синдром (сахарный диабет, снижение веса, холелитиаз, стеаторея и гипохлоргидрия) возникает не во всех случаях. Иногда соматостатиномы могут проявляться симптомами карциноидного синдрома, синдрома Кушинга или другими. В таких случаях диагноз ставится исключительно после ИГХ и электронно-микроскопического исследования – на основании экспрессии соматостатина в большинстве опухолевых клеток и наличия в них характерных эндокринных гранул. На момент постановки диагноза соматостиномы ПЖ обычно достигают больших размеров, дают метастазы в печень и имеют крайне плохой прогноз.
Нефункционирующие НЭО ПЖ (вернее опухоли без выраженного клинического синдрома) – самые сложные для постановки диагноза опухоли. Определить степень нейроэндокринной дифференцировки этих опухолей позволяет только экспрессия в них общих маркеров нейроэндокринной дифференцировки – синаптофизина, хромогранина А [1, 11, 14, 15]. НЭО ПЖ чаще всего метастазируют в печень и лимфатические узлы (парапанкреатические, ворот печени, парааортальные, мезентериальные), в редких случаях – в лимфатические узлы средостения и подмышечные, еще реже в кости, брюшину, легкие, почки, щитовидную железу. Примерно 90% пациентов с НЭО ПЖ, у которых не выявлены метастазы, и около 50% пациентов с выявленными метастазами живут 5 и более лет.
Факторами более благоприятного прогноза и длительного выживания пациентов с высокодифференцированными злокачественными НЭО ПЖ, по мнению Chu Q.D. et al. [16], являются: радикальное удаление опухоли; отсутствие метастазов в печень или появление их на поздних стадиях прогрессирования опухоли; применение «агрессивной» ХТ (в случае наличия метастазов в печени).
Факторы неблагоприятного прогноза всех высокодифференцированных НЭО ПЖ: нерадикальность выполненной операции; наличие метастазов в печень; низкая степень дифференцировки клеток опухоли; инвазия кровеносных (рис. 2 А), лимфатических сосудов и нервов (рис. 2 Б) (что наблюдается в 90% опухолей с метастазами или массивной инвазией окружающих органов и тканей и лишь в 30% опухолей без метастазов). Самыми важными факторами плохого прогноза высокодифференцированных НЭО ПЖ являются метастазы в печень, высокий митотический индекс и индекс Ki67. Определение индекса мечения Ki67 НЭО ЖКТ и ПЖ в соответствии с рекомендациями ENETS является золотым стандартом при определении риска прогрессирования НЭО ПЖ, выборе лечения и оценке эффективности ХТ (17). По нашим данным, более злокачественный потенциал и высокий риск появления метастазов в печень определяет и иммунофенотип клеток НЭО ПЖ. Так, факторами плохого прогноза является ко-экспрессия маркеров нейроэндокринной (хромогранина А, синаптофизина) и экзокринной дифференцировки (маркера клеток протокового эпителия – цитокератина 19, эпителиально-мембранного антигена, муцинов) (рис. 3 А и Б), а также ко-экспрессия нескольких гормонов в одной и той же клетке опухоли (например, инсулина и гастрина, инсулина и соматостатина, соматостатина и кальцитонина, АКТГ, гастрина и т. д.) [7]. Показано также, что экспрессия ассоциированного с метастазами гена 1 (MTA1) является еще одним потенциальным маркером злокачественного потенциала НЭО ПЖ [18].
НЭО пищевода. Высокодифференцированные НЭО пищевода встречаются очень редко (примерно 0,05% всех НЭО ЖКТ). Обычно они имеют крупные размеры (более 4 см), локализуются в дистальном отделе пищевода и не дают специфических гормональных синдромов.
НЭО желудка. Опухоли 1 типа – самые многочисленные из высокодифференцированных НЭО желудка (табл. 3). Они локализованы в пределах слизистого и подслизистого слоев тела желудка, образованы чаще всего ECL-клетками, продуцирующими гистамин, и обычно ассоциируются с развитием эндокринных синдромов. НЭО желудка 2 типа встречаюся реже, чем 1 типа, но могут развиваться при синдроме МЭН-1 и сопровождаться симптомами синдрома Золлинера-Эллисона (ЗЭС). Опухоли небольших размеров могут быть удалены эндоскопически и имеют хороший прогноз. НЭО желудка 3 типа – это обычно опухоли из ECL-, серотонин- или гастрин-продуцирующих клеток, без преимущественной локализации в желудке. Опухоли 2 и 3 типа размером более 2 см, с инвазией сосудов, глубокой инвазией мышечной пластинки обычно дают метастазы в лимфатические узлы [19, 20].
НЭО двенадцатиперстной и тощей кишки. Высокодифференцированные НЭО и локализованные в двенадцатиперстной и тощей кишке обычно растут бессимптомно [15]. Выделяют 4 типа НЭО: гастриномы, соматостатиномы, нефункционирующие серотонин-, гастрин- или кальцитонин-продуцирующие нейроэндокринные карциномы и ганглиоцитарные параганглиомы. До 2/3 опухолей этой локализации составляют гастриномы, которые обычно локализуются в верхних отделах двенадцатиперстной кишки, ассоциированы с СЗЭ, имеют небольшие размеры (не более 1 см), могут быть спорадическими, а при синдроме МЭН-1 – множественными. Дуоденальные гастриномы даже небольших размеров (менее 0,5-1 см) могут давать метастазы в регионарные лимфатические узлы, которые иногда значительно больше первичной опухоли, поэтому в некоторых случаях их ошибочно трактуют как первичные гастриномы ПЖ или «первичные гастриномы лимфатических узлов». Дуоденальные соматостатиномы обычно локализуются в области Фатерова соска и не вызывают специфического синдрома. При инвазии мышечного слоя вероятность появления метастазов в регионарных лимфатических узлах очень велика. Дуоденальные ганглиоцитарные параганглиомы обычно локализуются вблизи Фатерова соска и даже при размерах более 2 см и инвазии мышечной пластинки имеют благоприятный прогноз.
Читайте также: