Нарушение метаболизма мочевины на МРТ головного мозга
Добавил пользователь Alex Обновлено: 29.01.2026
Диагностика глутаровой ацидурии 1 типа по КТ, МРТ головного мозга
а) Терминология:
1. Сокращения:
• Глутаровая ацидемия 1 типа (ГА1)
• Недостаточность митохондриальной глутарил-КоА-дегидрогеназы (ГКДГ)
2. Определение:
• Врожденное нарушение метаболизма, характеризующееся энцефалопатическими кризами и приводящее к развитию дистонически-дискинетических двигательных нарушений
б) Визуализация:
1. Общие характеристики глутаровой ацидурии 1 типа:
• Лучший диагностический критерий:
о Расширенные сильвиевы борозды и гиперинтенсивные на Т2-ВИ/FLAIR базальные ганглии
• Локализация:
о Сильвиевы борозды, БГ
• Размеры:
о Расширение сильвиевых борозд
• Морфология:
о Малые размеры лобных и теменных покрышек → расширение сильвиевых борозд по типу крыльев летучих мышей
2. КТ признаки глутаровой ацидурии 1 типа:
• Бесконтрастная КТ:
о У >95% пациентов наблюдается кистозное расширение ликворных пространств средней черепной ямки:
- Расширение сильвиевых борозд (93%), расширение цистерны среднего мозга (86%)
- Гипоплазия лобных/височных покрышек, которые в норме покрывают островки
о Снижение плотности полосатых тел
о Развитие макроцефалии на ранних стадиях, развитие атрофии на поздних (проявляется преимущественно расширением желудочков)
о Возникновение субдуральной гематомы при минимальном травмирующем воздействии
• КТ с контрастированием:
о Накопление контраста отсутствует
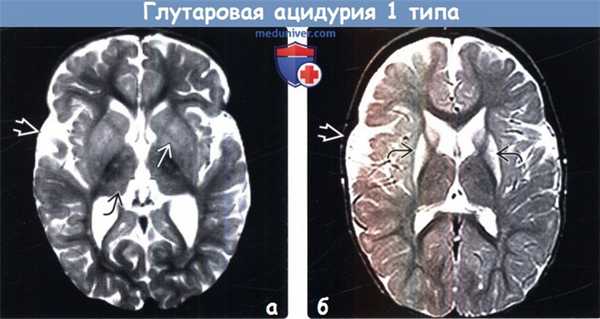
(а) МРТ, Т2-ВИ, аксиальный срез: определяется легкое расширение сильвиевых борозд и ранние изменения сигнала со стороны базальных ганглиев BE и таламусов, имеющих пестрый внешний вид. Наличие макрокрании -дополнительный признак, помогающий в постановке диагноза, когда другие изменения при глутаровой ацидурии 1 типа (ГА1) выражены минимально.
(б) МРТ-исследование ребенка в возрасте 21 месяца через длительный промежуток времени после разрешения метаболического криза, Т2-ВИ, аксиальный срез: выявляется атрофия легкой степени выраженности, глиоз базальных ганглиев, также сохраняется расширение сильвиевых борозд.
3. МРТ признаки глутаровой ацидурии 1 типа:
• Т1-ВИ:
о Кистозно расширенные ликворные пространства сильвиевых борозд изоинтенсивны по отношению СМЖ:
- Возможно уменьшение их размеров со временем
о Субэпидемиальные псевдокисты (исчезают в течение шести месяцев)
о Гипоплазия мозговой ткани лобно-теменных областей
о Задержка миелинизации
о Иногда паттерн архитектоники извилин может выглядеть слегка незрелым
• Т2-ВИ:
о Обычный: ↑ интенсивности сигнала от хвостатых ядер/скорлупы > бледного шара
о Иногда: изменения сигнальных характеристик бледных шаров и зубчатых ядер может наблюдаться даже вне кризов:
- Может предшествовать появлению изменений со стороны хвостатого ядра и скорлупы
о Развитие с течением времени атрофии полосатых тел
о При тяжелой форме: возможно вовлечение белого вещества (БВ), таламусов, зубчатых ядер
• FLAIR:
о Изменения идентичны таковым на Т2-ВИ
• ДВИ:
о Острая фаза: огранич:ение диффузии в области БГ и отдельных трактах БВ; изменений по сравнению с данными КТ- или других МР-томограмм могут наблюдаться чаще
• Постконтрастные Т1-ВИ:
о Контрастное усиление отсутствует
• МР-спектроскопия:
о ↑ холина/Cr, ↓ пика NAA
о Во время криза: ± повышение пика лактата
4. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ
• Совет по протоколу исследования: О МР-спектроскопия, ДВИ
в) Дифференциальная диагностика глутаровой ацидурии 1 типа:
1. Неслучайная травма:
• Гипоплазия покрышек может имитировать атрофию
• ГА1 не вызывает переломы
• При ГА1 субдуральная гематома (СДГ) обусловлена разрывом мостовых вен на фоне расширения ликворных пространств, атрофии
• При ГА1 в условиях отсутствия расширения ликворных пространств СДГ не возникает
• Травма головы = наиболее частая причина смерти:
о СДГ-наиболее частая находка, часто имеет межполушарную локализацию
о Переломы черепа с субарахноидальным и эпидуральным кровоизлияниями
о Отек мозга, ушиб(ы), повреждения вследствие воздействия сдвигающей силы
2. Другие патологические состояния с двусторонним кистозным расширением ли:кворных пространств средней черепной ямки:
• Мукополисахаридозы:
о Типы 1-4: Гурлер, Хантера, Санфилиппо, Шейе, Марото-Лами, Слая
о Отложение в твердой мозговой оболочке СМЖ-подобного мукополисахарида при всех типах, кроме синдрома Моркио (4 типа)
• «Идиопатические» арахноидальные кисты средней черепной ямки:
о В 5% могут быть двусторонними, обычно бессимптомны
о Ликворная интенсивность сигнала; на FLAIR может слегка отличаться от ликвора
о На ДВИ ограничение диффузии отсутствует
3. Другие причины макроцефалии:
• Гидроцефалия:
о Врожденная, посттравматическая или обструктивная
о Непропорциональное ширине борозд расширение желудочков
о Расширение височных рогов боковых желудочков, закругление передних рогов боковых желудочков, трансепендимальный ток СМЖ
• Идиопатическое расширение субарахноидальных пространств (САП) в течение 1 -го года жизни
• Доброкачественная семейная макроцефалия:
о Тенденция к крупным размерам головы у членов семьи
(а) MPT, FLAIR, аксиальный срез: у семимесячного ребенка определяется типичное расширение сильвиевых борозд. Отмечается ожидаемый аномальный сигнал от бледных шаров, а также от белого вещества больших полушарий. Последнее является необычным проявлением и, вероятно, отражает комбинацию у этого ребенка глутаровая ацидурия 1 типа (ГА1) с недостаточностью дигидропте-ридинредуктазы (ДГПР).
(б) МРТ, Т2-ВИ: у этого же ребенка определяются изменения белого вещества диффузного характера, а также аномальный сигнал от бледных шаров и таламусов.
г) Патология:
1. Общие характеристики глутаровой ацидурии 1 типа:
• Этиология:
о ГКДГ необходима для метаболизма лизина, гидроксилизина и триптофана
о ↑ ГКДГ → накопление глутаровой, глутаконовой и 3-ОН-глутаровой кислоты
о Накапливаемые вещества токсичны для клеток полосатых тел и белого вещества
• Генетика:
о Аутосомно-рецессивное наследование
о Мутации гена GCDH (Chr 19р13.2) приводят к замене аминокислот в белке
о Различные мутации обусловливают различные клинические проявления:
- Европейский вариант (наиболее частый): Arg402Trp
- Варианту амишей, чувствителен к рибофлавину: Ala421Val
- Тяжелый вариант, остаточная ферментативная активность - 1%, сохранение симптомов несмотря на лечение: Glu365Lys
о Редко наблюдается манифестация во взрослом возрасте: сложный гетерозиготный характер с делецией и новой миссенс-мутацией
• Ассоциированные аномалии:
о Эмбриология: токсические воздействия во внутриутробном развитии препятствуют оперкулизации, происходящей в третьем триместре
о Легкая печеночная недостаточность во время криза
2. Стадирование и классификация:
• Симптоматическая стадия: лобно-височная атрофия, изменения сигнала от БГ
• Пресимптоматическая стадия: отсутствие симптомов и изменений со стороны БГ, но имеет место расширение ликворных пространств
3. Макроскопические и хирургические особенности:
• Макрокрания, лобно-височная атрофия/гипоплазия; ↑ ликворных пространств ± СДГ
• Гипо-и демиелинизация
4. Микроскопия:
• Вакуолизация и расщепление миелина, избыточное количество внутримиелиновой жидкости
• Изменения по типу спонгиоза, потеря нейронов в БГ

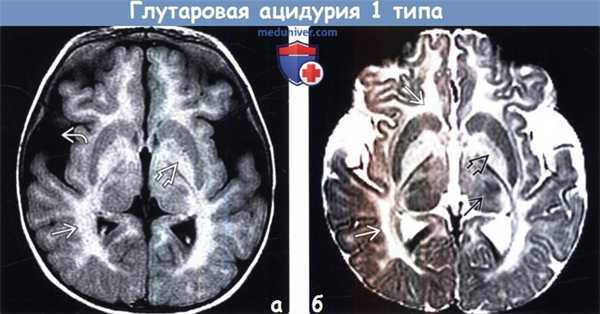
(а) МРТ в динамике, ДВИ: у этого же ребенка в возрасте почти двух лет определяется огра -ничение диффузии, подтверждающее активность процесса и явное прогрессирование изменений, особенно со стороны белого вещества. Также отмечается ограничение диффузии и в области вовлеченных в процесс бледных шаров.
(б) МРТ, Т2-ВИ, полученное в рамках того же исследования, что и ДВИ: определяется выраженное прогрессирование изменений со стороны белого вещества. Белое вещество теперь изо-интенсивно СМЖ. Расширенные сильвиевы борозды остаются весьма характерным для глутаровой ацидурии 1 типа (ГА1) признаком.
г) Клиническая картина:
1. Проявления глутаровой ацидурии 1 типа:
• Наиболее частые признаки/симптомы:
о Изначально отмечается нормальное развитие
о Острая энцефалопатия, судороги, дистония, хореоатетоз, умственная отсталость
• Острое начало отмечается у большинства пациентов:
о Эпизодические кризы следуют за триггером (инфекция, иммунизация, хирургия):
- Острая Рея-подобная энцефалопатия, кетоацидоз, ↑ NH4+, рвота
- Дистония, опистотонус, судороги, гипергидроз
- При наблюдении в динамике: тревожность ребенка (сохранность интеллекта >> моторные нарушения); быстрый рост головы у младенца → выступание лобных бугров; тяжелая дистония
• Скрытое начало (25%): дистония без кризов
• Пресимптоматическая стадия может оставаться бессимптомной: диагностика, лечение, исключение катаболического стресса
• Редкая бессимптомная в условиях отсутствия лечения форма: лобно-теменная атрофия присутствует, однако БГ в норме
• Диагностика: часто между манифестацией и постановкой диагноза проходит длительный промежуток времени:
о Тандемная масс-спектрометрия образцов крови новорожденного на карточке-фильтре:
- Хроматография с масс-спектрометрией; спектроскопия мочи
о Недостаточность или отсутствие активности ГКДГ в фибробластах
о Данные лабораторных методов исследования (в периоды между кризами могут быть относительно в норме):
- Метаболический ацидоз/кетоз, гипогликемия, ↓ карнитина
- Органические кислоты в моче: ↑ глутаровой, глутаконовой и 3-ОН-глутаровой кислот
2. Демография:
• Возраст:
о Обычно манифестирует в течение первого года жизни
• Половая принадлежность:
о Не выражена
• Этническая принадлежность:
о Частота носительства у амишей старого обряда - 10%
• Эпидемиология:
о 1:30000 новорожденных
3. Течение и прогноз:
• Симптоматическая стадия: у большинства пациентов серьезные нарушения, 20% умирают в возрасте до пяти лет
• Пресимптоматическая стадия: у многих (не всех) заболевание остается бессимптомным при своевременной диагностике и терапии
• Начало лечения до первого энцефалопатического криза, исключение катаболических кризов могут улучшить исход
• Неблагоприятный прогноз при поступлении пациента в лечебное учреждение с энцефалопатическим кризом
4. Лечение:
• Постановка диагноза до рождения возможна:
о Анализ ДНК: культивированные клетки амниотической жидкости и биопсия ворсинок хориона О УЗИ и МРТ плода: расширение перисильвиевых ликворных пространств в третьем триместре беременности
• Раннее начало лечения может предотвратить развитие или облегчить симптомы и изменения при диагностической визуализации:
о Низкобелковая диета (снижение потребления триптофана и лизина), напиток из синтетических белков
о Прием рибофлавина (витамин В2) с целью снабжения кофактором для ГКДГ
о Замена перорально принимаемого карнитина; гамма-аминомасляная кислота (ГАМК), аналог (баклофен)
д) Диагностическая памятка. Советы по интерпретации изображений:
• Предполагайте глутаровую ацидурию (ГА1) у детей младшего возраста с двусторонним расширением сильвиевых борозд и изменениями со стороны базальных ганглиев
• Изменения сигнальных характеристик от бледных шаров и зубчатых ядер может наблюдаться даже вне криза; могут предшествовать вовлечению в процесс хвостатых ядер и скорлупы
Нарушение метаболизма мочевины на МРТ головного мозга
Глутаровая ацидурия 1 типа на МРТ головного мозга
а) Терминология:
• Глутаровая ацидемия 1 типа (ГА1), недостаточность митохондриальной глутарил-КоА-дегидрогеназы (ГКДГ)
• Врожденное нарушение метаболизма, характеризующееся энцефалопатическими кризами и приводящее к развитию дистонически-дискинетических двигательных нарушений
б) Визуализация глутаровой ацидурии 1 типа:
• Расширенные сильвиевы борозды (вследствие гипоплазии лобных/височных покрышек) и гиперинтенсивные на T2-BM/FLAIR базальные ганглии
о Часто: ↑ интенсивности сигнала от хвостатых ядер/скорлупы > бледного шара
о Иногда: изменения сигнальных характеристик бледных шаров и зубчатых ядер может наблюдаться даже вне кризов
о Тяжелая форма: возможно вовлечение белого вещества, таламусов, зубчатых ядер
о Выраженная атрофия базальных ганглиев на поздних стадиях заболевания
• Изменения имитируют таковые при жестоком обращении с детьми: мостовые вены, проходящие в расширенных ликворных пространствах, легко подвергаются разрыву → субдуральные гематомы
(а) На рисунке аксиального среза изображена характерная для глутаровой ацидурии 1 типа (ГА1) картина изменений. Сильвиевы борозды расширены, сигнал от базальных ганглиев диффузно и симметрично изменен.
(б) МРТ, Т2-ВИ, аксиальный срез: у ребенка в возрасте семи месяцев определяется расширение сильвиевых борозд. Обратите внимание на отек и аномальное двустороннее повышение интенсивности сигнала от базальных ганглиев включая головки хвостатых ядер, скорлупу и бледные шары. Также отмечается задержка миелинизации. (а) МРТ, ДВИ, аксиальный срез: у младенца в разгар тяжелого метаболического криза наблюдается, что головки хвостатых ядер и скорлупа билатерально имеют гиперинтенсив -ный сигнал вследствие ограничения диффузии.
(б) Карта ИКД: отмечается снижение интенсивности сигнала от тех же структур - хвостатых ядер и скорлупы, что подтверждает ограничение диффузии, вызванное острым поражением, а не эффектом Т2-просвечивания.
в) Дифференциальная диагностика:
• Неслучайная травма
• Патологические состояния с двусторонним кистозным расширением ликворных пространств средней черепной ямки:
о Мукополисахаридозы
о «Идиопатические» арахноидальные кисты средней черепной ямки
• Другие причины макроцефалии:
о Гидроцефалия
о Идиопатическое расширение субарахноидальных пространств (САП) в течение 1-го года жизни
о Доброкачественная семейная макроцефалия
г) Клиническая картина глутаровой ацидурии 1 типа:
• Развитие эпизодических кризов обусловлено действием триггера (инфекция, иммунизация, хирургическое вмешательство)
• После каждого криза происходит увеличение степени повышения интенсивности сигнала/атрофии базальных ганглиев
• Обычно проявляется в течение первого года жизни
• Частота носительства у амишей старого обряда - 10%
а) Терминология:
• 6 вариантов нарушения цикла метаболизма мочевины: о Недостаточность орнитинтрансаминазы (ДОТА)
о Недостаточность карбамоилфосфатсинтетаз 1
о Цитруллинемия или недостаточность аргининсукцинатсинтетазы
о Аргининосукцинатная ацидурия или недостаточность аргинин-сукцинатлиазы
о Аргининемия или недостаточность аргиназы (AD)
о Недостаточность N-ацетилглутаматсинтетазы
б) Визуализация нарушений метаболизма мочевины:
• Новорожденные: глубокие ядра, на глубине борозд коры лобной, теменной и островковой > височной долей
• Более старший возраст: та же локализация или асимметричные изменения в коре/субкортикальном белом веществе, имитирующие инсульт
• Сохранность структур задней черепной ямки
• Острая/подострая стадия: ↑ интенсивности сигнала на Т2-ВИ, отек вовлеченных в процесс областей
• Острая/подострая стадия: изо-/гиперинтенсивный сигнал на ДВИ, изо-/гипоинтенсивный сигнал на картах ИКД
• Острая/подострая стадия: ↓ пика миоинозитола (Ml), ↑ пика глутамин-глутамата (glx), ↑ липиды/лактат
(а) МРТ, Т2-ВИ, аксиальный срез: у новорожденного мальчика возрастом два дня с острыми проявлениями недостаточности орнитинтрансаминазы (ДОТА) определяется аномальное повышение интенсивности сигнала между латеральными ядрами бледных шаров и скорлупой.
(б) МРТ, ДВИ, аксиальный срез: у этого же пациента определяется ограничение диффузии (аномальное повышение интенсивности сигнала) в области тех же анатомических структур: между бледными шарами и скорлупой, распространяясь в хвостатые ядра: данные изменения сопровождаются соответствующим снижением ИКД. Также наблюдается менее выраженное повышение интенсивности сигнала от таламусов (со снижением ИКД). (а) Протонная МР-спектроскопия (ТЕ = 144 мс): у новорожденного с острыми проявлениями определяется два инвертированных дублета лактата (на 1,33 ppm) и пик 1,2-пропендиола (обнаруживается в противосудорожных средствах, на 1,1 ppm). Кроме того, отмечается высокий пик глутамин-глутамата В (glx) на 2,1-1,4 ppm.
(б) МРТ, FLAIR, корональный срез: у другого ребенка с ДОТА в хронической стадии определяется повышение интенсивности сигнала от коры и субкортикального белого вещества задних отделов инсулярных и височно-теменных областей, что наиболее заметно на глубине борозд.
в) Дифференциальная диагностика:
• Гипоксически-ишемическая энцефалопатия
• Артериальный ишемический инсульт
• Митохондриальные заболевания
• Органические ацидемии
• Некетотическая гиперглицинемия
г) Патология:
• Процессы цикла метаболизма мочевины: азот → мочевина → моча; цикл предотвращает накопление токсичных азотсодержащих продуктов
• ↑ аммиака → ↑ глутамата → ↑ глутамина в астроцитах → отек + дисфункция
д) Клиническая картина:
• Триада: гипераммонемия, энцефалопатия и респираторный алкалоз
Диагностика нарушения обмена мочевины по КТ, МРТ головного мозга
а) Определение:
• Шесть вариантов нарушения цикла метаболизма мочевины:
о Недостаточность орнитинтрансаминазы (ДОТА)
о Недостаточность карбамоилфосфатсинтетазы 1
о Цитруллинемия или недостаточность аргининсукцинатсинтетазы
о Аргининосукцинатная ацидурия или недостаточность аргинин-сукцинатлиазы
о Аргининемия или недостаточность аргиназы (AD)
о Недостаточность N-ацетилглутаматсинтетазы
1. Общие характеристики нарушения обмена мочевины:
• Лучший диагностический критерий:
о Новорожденный с появлением в течение 24-48 часов отека базальных ганглиев (БГ) и ↑ интенсивности сигнала на ДВИ от коры
• Локализация:
о Новорожденные: глубокие ядра, на глубине борозд коры лобной, теменной и островковой > височной долей
о Более старший возраст: та же локализация или асимметричные изменения в коре/субкортикальном белом веществе (БВ), имитирующие инсульт
о Задняя ямка спасена
2. КТ признаки нарушения обмена мочевины:
• Бесконтрастная КТ:
о ↓ плотности глубоких ядер, БВ + коры с их отеком → атрофия в хроническую стадию
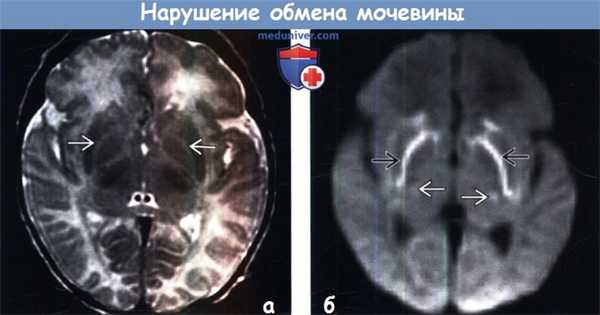
(а) МРТ, Т2-ВИ, аксиальный срез: у новорожденного мальчика возрастом два дня с острыми проявлениями недостаточности орнитинтрансаминазы (ДОТА) определяется аномальное повышение интенсивности сигнала между латеральными ядрами бледных шаров и скорлупой.
(б) МРТ, ДВИ, аксиальный срез: у этого же пациента определяется ограничение диффузии (аномальное повышение интенсивности сигнала) в области тех же анатомических структур: между бледными шарами и скорлупой, распространяясь в хвостатые ядра: данные изменения сопровождаются соответствующим снижением ИКД. Также наблюдается менее выраженное повышение интенсивности сигнала от таламусов (со снижением ИКД).
3. МРТ признаки нарушения обмена мочевины:
• Т1-ВИ:
о Подострая/хроническая стадия: ↑ интенсивности сигнала от вовлеченных в процесс коры, глубоких ядер
• Т2-ВИ:
о Острая/подострая стадия: ↑ интенсивности сигнала, отек вовлеченных в процесс областей
о Хроническая стадия: потеря объема, глиоз- кистозное изменение
• ДВИ:
о Острая/подострая стадия: изо-/гиперинтенсивный сигнал на ДВИ, изо-/гипоинтенсивный сигнал на картах ИКД
• МР-спектроскопия:
о Острая/подострая стадия: ↓ пика миоинозитола, ↑ пика глутамин-глутамата, ↑ липиды/лактат
4. Рекомендации по визуализации:
• Лучший инструмент визуализации: МРТ
• Совет по протоколу исследования: Т1 -ВИ, Т2-ВИ, ДВИ, МР-спектроскопия
в) Дифференциальная диагностика нарушения обмена мочевины:
1. Гипоксически-ишемическая энцефалопатия:
• Латеральные отделы скорлупы и вентролатеральный таламус; трудно дифференцировать в хроническую стадию
2. Артериальный ишемический инсульт:
• Соответствие локализации изменений бассейну кровоснабжения
3. Метаболические нарушения:
• Митохондриальные заболевания
• Органические ацидемии: поражение бледных шаров, но не коры; метаболический ацидоз/кетоз
• Некетотическая гиперглицинемия: отсутствие вовлечения в процесс базальных ганглиев
1. Общие характеристики нарушения метаболизма мочевины:
• Этиология:
о Процессы цикла метаболизма мочевины: азот → мочевина → моча, цикл предотвращает накопление токсичных азотсодержащих продуктов
о ↑ аммиака → ↑ глутамата → ↑ глутамина в астроцитах → отек + дисфункция
• Генетика:
о Все варианты нарушений, за исключением ДОТА (Х-сцепленное) имеют аутосомно-рецессивный тип наследования
2. Макроскопические и хирургические особенности:
• Отек головного мозга в острую стадию; атрофия + улегирия в хроническую стадию
3. Микроскопия:
• Астроциты Альцгеймера 2 типа в СВ; спонгиоз СВ, БВ
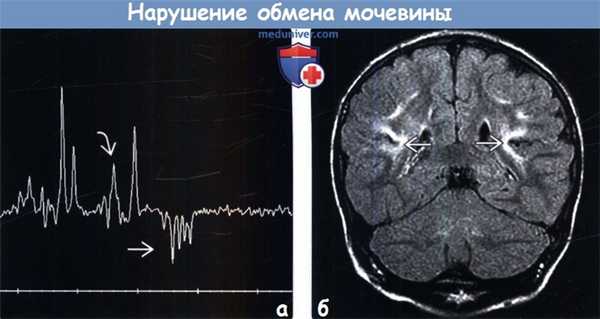
(а) Протонная МР-спектроскопия (ТЕ = 144 мс): у новорожденного с острыми проявлениями определяется два инвертированных дублета лактата (на 1,33 ppm) и пик 1,2-пропендиола (обнаруживается в противосудорожных средствах, на 1,1 ppm). Кроме того, отмечается высокий пик глутамин-глутамата В (glx) на 2,1-1,4 ppm.
(б) МРТ, FLAIR, корональный срез: у другого ребенка с ДОТА в хронической стадии определяется повышение интенсивности сигнала от коры и субкортикального белого вещества задних отделов инсулярных и височно-теменных областей, что наиболее заметно на глубине борозд.
д) Клиническая картина:
1. Проявления нарушения метаболизма мочевины:
• Наиболее частые признаки/симптомы:
о Триада: гипераммонемия, энцефалопатия и респираторный алкалоз
о Прогрессирующая апатия, гипотермия, рвота, апноэ
о У новорожденных энцефалопатия развивается в течение >24-48 часов
о Эпизодичческий характер у пожилых пациентов (часто при ↑ потребления белка или ↑ катаболизма)
• Клинический профиль:
о ↑ уровня аммиака в крови (за исключением болезни Альцгеймера)
о Диагноз: оценка ферментатов клеток печени/ДНК
2. Демография:
• Возраст:
о Новорожденные - тяжелое течение, лица более старшего возраста - менее тяжелое течение
• Эпидемиология:
о Белые > афроамериканцы
о ДОТА чаще
3. Течение и прогноз:
• При лечении становятся более благоприятными, но у большинства пациентов развивается умственная отсталость
• Новорожденные: более неблагоприятный прогноз с более высокой смертностью
4. Лечение:
• Гемодиализ при остром кризе
• Пересадка печени при тяжелой форме
• ↓ потребление белка, адекватное потребление калорий, биологически активные добавки
• Бензоат натрия/фенилбутират/фенилацетат
• Вальпроат противопоказан; может привести к летальному исходу
Центральный понтинный миелинолиз
Центральный понтинный миелинолиз — это острая ограниченная невоспалительная демиелинизация в средней части основания моста мозга. Возникает в результате неправильной коррекции гипонатриемии, развивающейся при нарушенной работе печени и почек, хроническом алкоголизме, некоторых гормональных расстройствах. Проявляется центральным тетрапарезом, псевдобульбарным синдромом, патологическими рефлексами. Для диагностики применяется МР-сканирование головного мозга, анализ спинномозговой жидкости, биохимические исследования крови. Лечение симптоматическое: назначаются кортикостероиды, инфузионные растворы, витамины, ноотропы.
МКБ-10
Общие сведения
Центральный понтинный миелинолиз (ЦПМ) — один из вариантов осмотического демиелинизирующего синдрома (ОДС). По данным МР-сканирования, характерные для ЦПМ нарушения определяются у 40-56 % пациентов, страдающих ОДС. В популяции патология встречается редко, еще реже она диагностируется при жизни: распространенность у неврологических больных составляет 0,4-0,56%. ЦПМ был впервые описан в 1959 г. у четырех пациентов с тетраплегией и псевдобульбарными расстройствами. В 70-х годах установлена связь заболевания с трансплантацией печени. Наследственной предрасположенности и статистически значимых половых различий в заболеваемости не обнаружено. Случаи заболевания у детей казуистичны.
Причины
Непосредственной причиной острой демиелинизации выступает быстрая медикаментозная коррекция гипонатриемии. В современной неврологии также обсуждается роль дисбаланса калия, фосфатов, магния в развитии ЦПМ. Однако эти предположения окончательно не подтверждены.
Болезнь не является специфическим синдромом, она может встречаться при заболеваниях, сопровождающихся расстройствами водно-солевого обмена. Типичные предпосылки к ее формированию:
- Поражения печени. Состояние зачастую выявляется у страдающих хронической печеночной недостаточностью, повреждениями органа на фоне хронического злоупотребления алкоголем. В редких случаях ЦПМ выступает как осложнение после трансплантации печени.
- Эндокринные расстройства. Предрасполагающими факторами являются сахарный диабет, аденома гипофиза. У женщин после родов острая ограниченная демиелинизация может быть одним из признаков болезни Шихана.
- Нарушения метаболизма. Патология нередко развивается у людей с тяжелой нутритивной недостаточностью, булимией. Важным предиктором формирования понтинного миелинолиза являются нарушения обмена мочевины.
- Прием диуретиков. При неправильном подборе дозы мочегонных врачом или склонности пациента к самолечению диуретиками происходят серьезные нарушения электролитного обмена, повышающие риск ЦПМ.
- Редкие причины. Иногда предпосылками выступают тяжелые вирусные инфекции, системная красная волчанка, иммунодефицитные состояния.
Патогенез
Согласно общепринятой гипотезе, для развития центрального миелинолиза необходимо снижение уровня натрия в крови менее 120 ммоль/л, продолжающееся более 48 часов. Для компенсации возникших изменений в организме активизируются два основных механизма. Первый из них заключается в выходе воды из клеток для баланса вне- и внутриклеточного содержания электролитов. Второй механизм — прекращение синтеза осмолитов (глутамата, таурина, глицина) в клетке.
Когда в стационаре проводится быстрая неконтролируемая коррекция содержания натрия, количество электролитов в интерстиции резко повышается, что в сочетании с увеличенным числом осмолитов во внеклеточной жидкости создает градиент концентрации. Для устранения этих нарушений внутриклеточная жидкость активно выходит наружу, а сами клетки сморщиваются и погибают. Такие патологические расстройства сохраняются в течение 2-3 суток от начала лечение гипонатриемии.
Вследствие изменений в центральных мозговых структурах возникает осмотический демиелинизирующий синдром (ОДС), которому больше всего подвержены мост, базальные ганглии, средний мозг. В патогенезе болезни также участвуют иммунные факторы — происходит формирование антител к миелиновой оболочке без развития воспаления. Центральный понтинный миелинолиз составляет около половины случаев ОДС, 10-23% приходится на экстрапонтинный миелинолиз (ЭПМ), в остальных случаях эти состояния сочетаются.
Патоморфологическая основа заболевания — распад миелиновых оболочек аксонов в центральных понтинных отделах мозга, который постепенно распространяется на все проводящие пути, кроме латеральных отделов. Однако сами нейроны не гибнут, поэтому потенциально ЦПМ можно рассматривать как обратимое состояние. В отличие от центральной формы патологии, при ЭПМ очаги демиелинизации расположены в ножках мозга, зрительном бугре, мозолистом теле.
Симптомы
В начальном периоде центрального понтинного миелинолиза клиническая картина соответствует проявлениям гипонатриемии: угнетение сознания, многократная рвота, судорожные приступы. После введения электролитных составов с натрием состояние больных быстро стабилизируется. Период благополучия длится не более 72 часов, однако в это время в структурах центральной нервной системы уже происходят патологические процессы.
Типичный вариант заболевания манифестирует очаговой неврологической симптоматикой. Основным признаком миелинолиза считается псевдобульбарный синдром, включающий дисфагию (нарушенное глотание), дисфонию (расстройство голосообразования), дизартрию. Наступает тотальный паралич — центральная тетраплегия, который становится причиной синдрома «запертого человека».
Расстройство проявляется повышением мышечного тонуса, гиперрефлексией, патологическими рефлексами орального, спинального автоматизма. При распространении демиелинизации на соседние участки мозга появляются гиперкинезы (атаксии, паркинсонизм, дистонии). К редким симптомам болезни относится вертикальный или горизонтальный парез взгляда, сходящееся косоглазие, понтинные альтернирующие синдромы (Мийяра-Гублера, Фовилля, Гасперини).
Осложнения
Основная опасность состояния состоит в необратимости процессов демиелинизации в головном мозге. Хотя при этом нейроны все ещё остаются живыми, медикаменты и методики, способные запустить процесс восстановления миелиновых оболочек проводящих путей, пока не разработаны.
Чем позже диагностирован ЦПМ, тем выше риск летального исхода или инвалидизирующих осложнений. Типичным последствием болезни является синдром «запертого человека», характеризующийся неспособностью реагировать на внешние стимулы из-за потери двигательной активности, речи. При этом у пациента в полной мере сохранено сознание.
Ранее показатель летальности при ЦПМ достигал 90-100%, но благодаря усовершенствованию медицинских протоколов успешность лечения таких больных значительно возросла. Смертность в основном наступает от присоединения соматических осложнений (пневмонии, уросепсиса). У 60-75% выживших сохраняются резидуальные проявления, зависящие от объема поражения мозга. Негативные последствия включают параличи, парезы, речевую дисфункцию, тяжелые когнитивные расстройства.
Диагностика
Опытным врачам-неврологам удается заподозрить патологию по сочетанию предыдущей гипонатриемии с признаками понтинного поражения мозга, однако вследствие тяжести состояния и неспецифичности симптомов своевременная диагностика болезни затруднена. Чтобы подтвердить наличие центрального понтинного миелинолиза, назначаются следующие методы исследования:
- МРТ головного мозга. На снимках обнаруживаются гиперинтенсивные (светящиеся) участки демиелинизации, симметричные очаги овальной формы в больших полушариях, сочетающиеся с повреждениями типа «бабочки» или «трезубца» в мосте. Для лучшей верификации демиелинизирующих процессов применяется Т2-режим МРТ.
- Электроэнцефалография. При ЭЭГ выявляется диффузное замедление биоэлектрической активности в обоих мозговых полушариях. При судорожных приступах исследование показывает патологическую бета-активность, наличие комплексов «острая-медленная волна».
- Анализ СМЖ. При исследовании цереброспинальной жидкости определяется повышение ликворного давления, мононуклеарный плейоцитоз, незначительное увеличение уровня белка. Для дифференциальной диагностики с нейроинфекциями выполняются серологические, бактериологические, вирусологические анализы.
- Биохимия крови. Для оценки дисэлектролитных изменений и наблюдения за динамикой состояния необходимо регулярно измерять количество натрия, калия, кальция. Поскольку поражение печени является распространенной причиной патологии, целесообразно делать печеночные пробы.
Лечение центрального понтинного миелинолиза
Специфическая терапия понтинного миелинолиза отсутствует. Пациентам назначается патогенетическое и симптоматическое лечение с учетом степени тяжести клинической картины, характера основной болезни, которая стала причиной гипонатриемии. Как один из вариантов лечения рассматривается повторная индуцированная гипонатриемия. Схемы лечения могут включать следующие фармацевтические средства:
- Кортикостероиды. Вероятно, препараты уменьшают вторичный воспалительный процесс в церебральной ткани, снижают риски осложнений. Они используются в комбинации с внутривенными иммуноглобулинами.
- Витамины. Препараты аскорбиновой кислоты, токоферола обладают антиоксидантной активностью. Применение витаминов группы В целесообразно для коррекции неврологического дефицита.
В периоде выздоровления необходима индивидуальная программа реабилитации, позволяющая устранить или уменьшить отдаленные последствия понтинного миелинолиза. Чтобы корректировать дизартрические нарушения, показаны занятия с логопедом. Для улучшения двигательной функции рекомендованы физиотерапия, механотерапия, лечебная физкультура. Хороший эффект отмечается после приема ноотропов, нейрометаболических препаратов.
Прогноз и профилактика
Центральный понтинный миелинолиз отличается неблагоприятным течением, высоким показателем смертности (до 12%). У выздоровевших пациентов, как правило, остаются резидуальные неврологические нарушения. Профилактика включает коррекцию стандартов оказания медицинской помощи, рациональное использование электролитных растворов, тщательное наблюдение за неврологическим статусом больных с гипонатриемией.
1. Центральный понтинный миелинолиз на фоне инфекции SARS-CoV-2 (клинические наблюдения)/ Воскресенская О.Н., Коваленко А.А., Надбитова Е.Б., Гринюк В.В., Климанов А.В., Шор Ю.М.// Неврология, нейропсихиатрия, психосоматика. — 2021. — №2.
2. Центральный понтинный и экстрапонтинный миелинолиз: обзор литературы и собственное наблюдение/ Е.М. Шевелева, Л.Г. Заславский, А.Г. Ковеленов, Е.А. Скорнякова// Ученые записки СПбГМУ им. акад. И.П. Павлова. — 2020. — №2.
Читайте также: