Нарушения метаболизма пиримидинов
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Как отмечалось выше, конечными продуктами метаболизма пуринов являются хорошо растворимые в воде соединения, такие, как аммиак, Р-аланин и пропионат. Вот почему при состояниях, характеризующихся избыточным образованием пиримидинов, клинические симптомы слабо выражены. В случаях гиперурикемии, обусловленной избыточной продукцией ФРПФ, наблюдаются повышенный синтез пиримидиновых нуклеотидов и соответственно увеличенная экскреция конечных продуктов метаболизма, в частности Р-аланина. Поскольку для синтеза тимидилата необходим нарушения метаболизма фолата и витамина В12 приводят к дефициту ТМР (влияние недостатка витамина В12 реализуется опосредованно).
Экскреция с мочой Р-аминоизобутирата—ауто-сомный рецессивный признак. Он распространен главным образом среди жителей Востока. Это нарушение
Таблица 35.3. Наследуемые нарушения метаболизма пиримидинов и связанные с ними изменения активности ферментов
не рассматривается как патологическое состояние (об этом уже шла речь при обсуждении катаболизма пиримидинов).
Описаны два типа первичной наследственной оротовой ацидурии. Более распространенная форма (хотя и встречающаяся весьма редко) связана с утратой во всех тестированных типах клеток функции двух ферментов — оротатфосфорибозилтрансферазы и оротидилат(ОМР)-декарбоксилазы (рис. 35.19). Организм пациентов с этим типом оротовой ацидурии можно считать ауксотрофным по пиримидину. Заболевание легко поддается лечению уридином. В детском возрасте для больных характерны отставание в развитии, мегалобластическая анемия и «оранжевая кристаллоурия» (оротовая кислота). При отсутствии лечения пиримидиновыми нуклеозидами пациенты подвержены инфекциям. Второй тип наследуемой оротовой ацидурии (тип II) связан с недостатком только ОМР-декарбоксилазы (рис. 35.19). У пациентов, страдающих оротовой ацидурией первого типа, основным продуктом экскреции является оротовая кислота. При обследовании больного ацидурией второго типа оказалось, что у него экскрети-руется главным образом оротидин и лишь небольшое количество оротовой кислоты. В эритроцитах пациентов с оротовой ацидурией типа I были значительно повышены удельные активности аспартаттранскарбамоилазы и дигидрооротазы; они, однако, возвратились к норме при пероральном приеме пациентами уридина. Эти наблюдения свидетельствуют о том, что конечные продукты пути биосинтеза пиримидинов регулируют активность рассматриваемых ферментов. В условиях недостатка конечных продуктов происходит дерепрессия, вероятно, координированного характера, по крайней мере этих двух ферментов.
Данные по энзимологии пути биосинтеза пиримидинов позволяют предполагать, что в одной белковой молекуле локализованы активные центры карбамоилфосфатсинтазы, аспартаттранскарбамоилазы и дигидрооротазы, а в другой — активные центры оротат-фосфорибозилтрансферазы и ОМР-декарбоксилазы.
У больных с недостаточностью орнитинтранс-карбамоилазы — митохондриального фермента клеток печени, ответственного за раннюю стадию синтеза мочевины и аргинина, наблюдается увеличение экскреции оротовой кислоты, урацила и уридина. По-видимому, в митохондриях этих пациентов в результате снижения активности орнитинтранскар-бамоилазы накапливается карбамоилфосфат. Митохондриальный карбамоилфосфат диффундирует в цитозоль, где используется как субстрат для синтеза пиримидиновых нуклеотидов de novo. При избыточном образовании оротовой кислоты диагностируется оротовая ацидурия. Обычно она проявляется в легкой форме и не сопровождается образованием кристаллов. Синтез и экскреция оротовой кислоты у таких больных усиливаются при употреблении в пищу продуктов богатых азотом, таких, как мясо.
Оротовую ацидурию могут вызывать по крайней мере два лекарственных препарата. Аллопуринол (4-гидроксипиразолопиримидин) - пуриновый аналог, ингибирующий ксантиноксидазу. Этот препарат широко используется для лечения некоторых форм подагры. Аллопуринол может фосфорибозилироваться оротат-фосфорибозилтрансферазой, конкурентно ингибируя фосфорибозилирование оротовой
Рис. 35.19. А. Ферментные дефекты и их последствия при оротовой ацидурии типа I, которая характеризуется недостаточностью двух ферментов — оротатфосфорибозилтрансферазы и оротидилат-декарбоксилазы. Б. Недостаточность оротидилат-декарбоксилазы, приводящая к оротовой ацидурии типа II. Пунктирные линии со знаком минус показывают пути ингибирования по принципу обратной связи в нормальных условиях. При оротовой ацидурии типа I с мочой экскретируется оротовая кислота. При оротовой ацидурии типа II в роли экскретируемых соединений выступают оротовая кислота и оротидин. Аббревиатуры расшифрованы на рис. 35.15.(Redrawn and reproduced, with permission, from Smith L. H. Ir. Pyrimidine metabolism in man. N. Engl. Med. 1973, 288, p. 764) кислоты. Более того, образующийся необычный нуклеотид подавляет оротидилатдекарбоксилазу, вызывая тем самым оротовую ацидурию и оротидинурию. Однако путь биосинтеза пиримидинов, по крайней мере у человека, «приспосабливается» к такому ингибированию, и «пиримидиновое голодание» имеет место только на ранних стадиях лечения препаратом.
6-Азауридин после превращения в 6-азауридилат функционирует как конкурентный ингибитор ОМР-декарбоксилазы, что приводит к увеличению экскреции оротовой кислоты и оротидина.
При специфическом поражении митохондрий клеток печени (синдром Рейе) имеет место вторичная оротовая ацидурия. Дело в том, что пораженные митохондрии оказываются неспособными утилизировать карбамоилфосфат, который, как и в случае наследуемой недостаточности орнитинтранскарбамоилазы, вызывает избыточное образование оротовой кислоты.
ЛИТЕРАТУРА
Ames В. N. et al. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis, Proc. Natl. Acad. Sci. USA, 1981, 78, 6858.
Henderson J. F. Regulation of Purine Biosynthesis, Monograph, No. 170, American Chemical Society, 1972.
Henderson J.F., Paterson A.R.P. Nucleotide Metabolism: An Introduction, Academic Press, 1973.
Jones M. Pyrimidine nucleotide biosynthesis in animal cells, Annu. Rev. Biochem., 1980, 49, 253.
Kempe T. D. et al. Stable mutants of mammalian cells that overproduce the first three enzymes of pyrimidine nucleotide biosynthesis, Cell, 1976, 9, 541.
Martin D. W. Jr., Gelfand E. W. Biochemistry of diseases of im-munodevelopment, Annu. Rev. Biochem., 1981, 50, 845.
Smith L.H. Jr. Pyrimidine metabolism in man, N. Engl. J. Med., 1973, 288, 764.
Stanbury J. B. et al. (eds.) The Metabolic Basis of Inherited Disease, 5th ed., McGraw-Hill, 1983.
Thelander L., Reichard P. Reduction of ribonucleotides, Annu. Rev. Biochem., 1979, 48, 133.
Wyngaarden J. B., Kelley W. N. Gout and Hyperuricemia, Gru-ne and Stratton, 1976.
Нарушения метаболизма пиримидинов
Пурины и пиримидины – органические вещества, входящие в структуру нуклеиновых кислот, коферментов и макроэргических соединений. Нарушение метаболизма азотистых оснований возникает вследствие генетически обусловленных дефектов ферментов, участвующих в обмене пуринов и пиримидинов.
Синонимы русские
Наследственные дефекты метаболизма азотистых оснований.
Синонимы английские
Purine and Pyrimidine Panel, Urine.
Метод исследования
Высокоэффективная жидкостная хроматография-масс-спектрометрия (ВЭЖХ-МС).
Единицы измерения
мкмоль / ммоль креатинина (микромоль на миллимоль креатинина).
Какой биоматериал можно использовать для исследования?
Разовую порцию мочи.
Как правильно подготовиться к исследованию?
- Исключить (по согласованию с врачом) прием мочегонных препаратов в течение 48 часов до сбора мочи.
Общая информация об исследовании
Пурины и пиримидины – гетероциклические органические азотистые вещества, входящие в состав нуклеотидов и нуклеозидов. Они являются важными структурными элементами нуклеиновых кислот (ДНК, РНК), источников энергии (например, АТФ), ферментов (НАДФ, НАД, ФАД).
Дефекты ферментов, принимающих участие в разных этапах метаболизма пуринов и пиримидинов, могут приводить к развитию заболевания. Патогенез данных состояний связан с накоплением в клетках и биологических жидкостях избыточного количества азотистых оснований и их метаболитов, которые могут быть токсичными и способными повреждать генетический материал и функцию клеток. Описано около 30 нарушений различных этапов метаболизма пуринов и пиримидинов, но клинически проявляются только 17. Основные лабораторные признаки данных заболеваний – это изменение содержания пуринов и пиримидинов в биологических жидкостях. Первые симптомы могут возникать как в раннем детстве, так и в старшем возрасте, а клинические проявления могут значительно варьироваться по степени тяжести. Наиболее часто при патологии пуринового и пиримидинового обмена повреждаются нервная система (задержка развития, аутизм, эпилептические приступы), кроветворная ткань (анемия) и почки (нефропатия, мочекаменная болезнь).
Классическим примером приобретенного нарушения пуринового обмена является подагра – заболевание, сопровождающиеся повышением уровня мочевой кислоты в крови и отложением уратов в тканях. Однако необходимо учитывать, что гиперурикемия (повышение уровня мочевой кислоты в крови) у людей старше 40 лет связана не только с генетической предрасположенностью, но и с особенностями питания, употреблением алкоголя, нарушением экскреторной функции почек. Гиперурикемия и подагра в более молодом или детском возрасте в большинстве случаев ассоциированы с наследственными дефектами ферментов пуринового обмена.
Тяжелое нарушение метаболизма пуринов – синдром Леша – Нихана (абсолютный дефицит гипоксантин-гуанин-фосфорибозилтрансферазы), который наследуется совместно с Х-хромосомой и проявляется тяжелыми неврологическими нарушениями, задержкой психомоторного развития, церебральным параличом, аутоагрессивным поведением и уратной нефропатией. Симптомы заболевания чаще становятся заметными в возрасте 3-12 месяцев. При данной патологии в биологических жидкостях возрастает концентрация мочевой кислоты и гипоксантина.
К врождённым нарушениям пуринового обмена относятся дегидроксиаденинурия, наследственная ксантинурия, синдром Келли – Зигмиллера и др. Дефицит аденозин-дезаминазы и пурин-нуклеозид-фосфорилазы приводит не только к неврологическим нарушениям, но и к снижению количества лимфоцитов и иммунодефициту, который проявляется рецидивирующими тяжелыми инфекциями.
Патология пиримидинового обмена наблюдается и диагностируется значительно реже. К нарушениям метаболизма пиримидинов относятся оротовая ацидурия, дефицит пиримидин-5-нуклеотидазы, дефицит дигидропиримидин-дегидрогеназы.
Некоторые из наследственных нарушений обмена азотистых оснований при своевременном выявлении поддаются коррекции, для других применяется симптоматическая терапия и разрабатываются новые методы лечения. Диагноз верифицируется на основании комплексных клинико-генеалогических данных и результатов лабораторного исследования.
«Диагностика нарушения обмена пуринов и пиримидинов в крови»
Цена: 4000 руб.
Материал: Кровь
Время забора: 7:00-12:00 сб. 7:00-11:00
Выдача результатов: до 8 рабочих дней
Условия подготовки к анализам:
Диагностика нарушения обмена пуринов и пиримидинов в крови
Пурины и пиримидины являются абсолютно незаменимыми компонентами клеток: они необходимы для синтеза ДНК и РНК, запасания и транспорта энергии, формирования коэнзимов и метаболитов синтеза фосфолипидов и углеводов, а также передачи сигнала в клетку и его преобразования. Учитывая такие разнообразные функции пуринов и пиримидинов, становится ясным, почему нарушения их обмена приводят к одновременному поражению сразу нескольких систем органов. Чаще эти нарушения обусловлены недостаточностью ферментов, необходимых для нормального метаболизма пуринов и пиримидинов, носят врожденный характер и проявляются в младенческом или раннем детском возрасте, хотя некоторые из них манифестируют уже во взрослом возрасте. На сегодняшний день известно около 30 таких нарушений, 17 из которых приводят к заболеваниям.
Несмотря на то что существуют некоторые закономерности в клинической картине этих врожденных болезней (например, частое поражение нервной системы и задержка умственного развития при нарушении обмена пиримидинов), клиническая картина многих из этих нарушений не имеет каких-либо специфических признаков. Все эти болезни могут быть очень похожи друг на друга. В связи с этим диагностировать эти заболевания, а тем более различить их между собой на основании только лишь симптомов невозможно. С другой стороны, своевременная диагностика этих заболеваний очень важна, так как она позволяет провести генетическое консультирование и в некоторых случаях начать терапию, которая может полностью избавить пациента от симптомов. Основным и единственным методом окончательной диагностики нарушений метаболизма пуринов и пиримидинов является лабораторный метод, в ходе которого одновременно исследуют пурины и пиримидины и все их метаболиты, известные на данный момент и значимые в клиническом плане.
В комплексное исследование входят следующие компоненты:
1. Метаболит синтеза пиримидинов – оротовая кислота. Дефект: наследственная оротовая ацидурия.
2. Пиримидиновые азотистые основания: цитозин, урацил (и его производное гидроксиметилурацил) и тимин. Дефект: наследственная тиминурацилурия.
3. Пиримидиновые нуклеозиды: цитизин, уридин (и его производное дезоксиуридин) и тимидин.
4. Метаболиты распада пиримидинов: дигидроурацил, уреидопропионовая кислота и β-аланин. Дефект: наследственный дефицит β-уреидопропионазы.
1. Пуриновые азотистые основания: гуанин и аденин.
2. Пуриновые нуклеозиды: гуанозин (и его производное дезоксигуанозин), аденозин (и его производное дезоксиаденозин) и инозин. Дефект: первичный иммунодефицит (недостаточность пуриннуклеозидфосфорилазы).
3. Метаболиты распада пуринов: ксантин и мочевая кислота. Дефект: синдром Леша – Нихана, тяжелый комбинированный иммунодефицит (недостаточность аденозиндезаминазы), ксантинурия.
В настоящий момент не существует более или менее четких указаний относительно того, в каких группах пациентов следует проводить исследование на пурины/пиримидины и их метаболиты. Учитывая разнообразную клиническую картину этих заболеваний, назначить этот анализ может практически любой специалист.
Ежегодно обнаруживают новые нарушения метаболизма пуринов и пиримидинов. В это комплексное исследование включены основные метаболиты, хотя все же не все. Поэтому нормальный результат этого исследования не может однозначно исключить наличие какого-либо заболевания из этой группы.
В некоторых случаях комплексный лабораторный анализ выявляет нарушения метаболизма пуринов и пиримидинов, которые, однако, никак не проявляются и клинического значения не имеют.
Следует отметить, что при обследовании пациента с подозрением на нарушение метаболизма пуринов и пиримидинов в большинстве случаев требуются дополнительные лабораторные тесты, с помощью которых удается оценить степень тяжести повреждения органов и тканей.
Нарушение метаболизма пиримидинов
Важнейшим нарушением пиримидинового обмена является оротовая ацидурия. Оротовая кислота – промежуточный продукт синтеза пиримидинов.
Оротовая ацидурия связана с блокадой метаболизма оротовой кислоты, нарушением синтеза уридина. Наследственный дефект ферментов, участвующих в декарбоксилировании и образовании кислоты (оротатфосфорибозилтрансферазы и оротидин – 5- фосфат-декарбоксилазы).
Признаки мегалобластической анемии (резистентной к терапии фолиевой кислотой и вит. В12).
Экскреция ортовой кислоты с мочой, в 1000 раз превышающая нормальное значение (возможна кристаллурия, закупорка мочеточника или уретры).
Отставание в росте и развитии (страдают быстрорастущие ткани).
ИД с преимущественным нарушением Т- клеточных ƒ.
Но, главным являются гематологические нарушения, т.к. усиленный синтез ДНК и РНК необходим для нормального гемопоэза.
Лечение: в течении всей жизни производными пиримидина.
Нарушение метаболизма пуринов
Нуклеиновые кислоты организма наполовину состоят из пуриновых нуклеотидов.
Основные проявления нарушения обмена пуринов:
Подагра (гиперурикемия и подагра самые распространенные).
Мочекислый инфаркт – при высокой гиперпродукции мочевой кислоты на фоне сопутствующей дегидратации или ограничения фильтрации.
Гипоурикемия – протекает бессимптомно.
а) ↓выработка МК (цирроз)
б) ↑выведения ее почками (метаболический дефект, в составе других заболеваний)
в) несбалансированное питание с перегрузкой глицином.
Отсутствие активности (тотальная недостаточность) гипоксантингуанинфосфорибазилтрансферазы. Наследование – сцепленное с Х-хромосомой. Фермент необходим для образования пуринов (катализирует реакцию реутилизации гипоксантина и гуанина).При рождении ребенок выглядит здоровым.
Патологические проявления: в течении первых месяцев отставание моторного развития, экстрапирамидные хореоатетоидные движения, гиперрефлексия. Взрывное агрессивное поведение (ребенок откусывает себе язык, кончики пальцев), в более старшем возрасте – тофи, артрит. Увеличение продукции мочевой кислоты (100-102 мг/л в сыворотке), увеличение экскреции с мочой, т.е. сопровождается клиническими признаками подагры. Болеют чаще мальчики от 6 до 16 лет. Склонность к самокалечению проявляется не ранее второго года жизни. Это представляет собой императивное желание (а не неспособность ощущать боль), ребенка сдерживают, т.к. могут возникать увечья. У этих больных обнаружено снижение активности ферментов синтеза дофамина в экстрапирамидной системе – но механизм заболевания до конца не ясен.
Подагра
Подагра – заболевание с нарушением пуринового обмена и накоплением мочевой кислоты в организме. Относятся к болезням «ошибок метаболизма». Распространенность – 2 % взрослого населения (США, Европы), чаще после 40 лет, чаще мужчины 95 %, у женщин – в климактерический период. Средне мировая частота ≈ 1,3 – 3,7 % (эстрогены оказывают антиурикемическое действие).
Подагра в переводе с греческого «нога в капкане». Tomas Sydenham сравнивал боль при подагре с болями от «зажима конечности в прессе». Известна врачам с древних времен. История ее насчитывает > 2 тыс. лет. В «Афоризмах» Гиппократа присутствует попытка различать подагру и др. заболевания суставов. Но классическое ее описание связывают с именем английского врача XVII века Сиденгамма («Трактат о подагре»).
У человека конечным продуктом метаболизма пуринов является мочевая кислота. Человек выводит около 1,5 гр. мочевой кислоты в день (60 % из эндогенных пуринов, 40% - из пищевых продуктов: икра, молока >100мг/100гр.), печень, почки, потроха, свежий судак, консервированная рыба, мясо (меньше, чем в рыбе), яйца, красное вино, темные сорта пива, кофе, какао, шоколад, чай. Молоко, сыры, крупы, фрукты, овощи – бедны пуринами. Мочевая кислота может быть образована только под действием фермента ксантиноксидазы (у человека этот фермент есть только в энтероцитах и печени). Мочевая кислота – слабо-кислый продукт, секретируется в крови, образует соли – ураты, выводится в основном почками (фильтрация, реабсорбция, секреция) 1/3 МК выводится через тонкий кишечник. Ураты легче выпадают в осодок при температуре < 37 о С (наиболее «холодным» суставом в организме человека является первый предплюснофаланговый сустав стопы).
Известно, что МК (3-тригидроксиксантин) и кофеин (3-метилксантин) являются структурными аналогами, поэтому существуют определенные представления о «допинговом» эффекте мочевой кислоты на умственную активность (энциклопедия Эфроимсон – гении – 40% носителей достоверных и высоко вероятных признаков гиперурикемии, хотя снижение МК не вызывает снижение интеллекта. Установлена связь между гиперурикемией и повышенной умственной активностью).
Среди заболеваний современного человека подагре принадлежит совершенно особое место – трудно найти другую болезнь, которая полностью сохранила бы свое первичное «лицо», несмотря на тысячелетнее существование.
Ускорить или спровоцировать подагрический криз может целый ряд факторов – физические перегрузки, травма (хирургические), эпизоды переедание, потеря жидкости (воздействие жаркого климата, сауны), алкогольный эксцесс, лекарства (главным образом диуретики, цитостатики), острая инфекция, инфаркт миокарда, стресс.
Г.А. Захарьин четко очерчивает круг лиц, у которых развивается или может развиться подагра: ''человек лет 40, хорошего сложения, излишнего питания и с хорошим пищеварением, хорошо кушающий и телесно недеятельный, давно уже замечающий у себя красную мочу, словом, кандидат на подагру, но не представляющий еще ни единого местного ее симптома''.
Диагностические признаки подагры: острый подагрический артрит с типичной его локализацией в области первого плюсневого сустава (у 60 % б-х), отложения в околосуставных тканях, хрящах, ушных раковинах, по ходу сухожилий мелоподобных пастообразных масс, тофусов, рентгенологические признаки – эрозивные изменения костных частей суставов, повышенный уровень мочевой кислоты в крови (гиперуринемия).
В настоящее время одним из эпидемиологически важных факторов, снижающих почечную экскрецию мочевой кислоты, признают избыточное накопление свинца в организме, особенно значимое для лиц с врожденной МК. В Англии издавна наблюдались четко документированные локальные эпидемии подагры, связанные с употреблением вина из Португалии (вино доставлялось в сосудах, которые покрывали составом свинца). Также эпидемии подагры отмечались в Австралии (колонисты использовали в качестве краски для домов свинцовый сурик), в США – во время «сухого закона» 20 х годов (свинец попадал в самодельный виски). В России – вышла «Белая книга», в которой приводятся данные о значительном загрязнении свинцом окружающей среды.
Подагра и гиперурикемия

В чем отличие подагры от простой гиперурикемии? Гиперурикемия — это лабораторный феномен, суть которого — увеличение содержания в крови мочевой кислоты. Не во всех случаях гиперурикемия приводит к возникновению клинически значимых симптомов. Подагра же — состояние, характеризующееся отложением кристаллов мочевой кислоты в тканях. Больше всего достается почкам и суставам. Подагры не бывает без гиперурикемии. Но как же последняя приводит к такому тяжелому заболеванию?
Мочевая кислота имеет свой предел растворимости. В чрезмерно кислой среде (c pH ниже 5,7) она выпадает в виде кристаллов уратных солей в мягких тканях. Оседая в суставах, соли мочевой кислоты вызывают чрезвычайно болезненные ощущения. Первым достается пальцам ног. Ураты для тканей — чужеродные соединения. В силу этого в тканях реализуется типовой патологический процесс — воспаление, которое в данном случае имеет скорее разрушающее, нежели защитное действие [1].
Но почему так происходит? Бывают случаи, когда мочевой кислоты образуется слишком много.
А) Повышенное образование пуринов и их усиленный распад (логичное следствие). Здесь причиной служит генетически детерминированная чрезмерная активность фосфорибозилпирофосфаттрансферазы (катализатор первой реакции синтеза пуринов), которая становится менее чувствительной к отрицательным регуляторным факторам — ГМФ и АМФ [6].
Б) Недостаточная реутилизация. Бывает так, что распадающиеся пурины утилизируются хуже, чем положено (не по плану) в силу дефектов гипоксантингуанинфосфорибозилтрансферазы (фермента, переводящего гипоксантин в ИМФ, а гуанин — в ГМФ). В результате мы получаем большое количество мочевой кислоты.
Ситуация может усугубляться, если с пищей поступает много нуклеиновых кислот, которые, распадаясь на пурины, превращаются в энтероцитах в мочевую кислоту и в таком виде всасываются в кровь. Алкоголь нарушает выведение мочевой кислоты (по пока неясным для меня самого причинам). При гиперурикемии противопоказаны диуретики (препараты, усиливающие выведение мочи, в особенности — из тиазидной группы), так как они повышают риск уратной нефропатии. Как видишь, гиперурикемия создает сплошные трудности. В отношении подагры надо сказать, что это инвалидизирующее заболевание, снижающее качество жизни и увеличивающее риск сердечно-сосудистых патологий [1].
Терапевтический подход
По вышеуказанным причинам первым вектором лечения подагры будет соблюдение диеты, которая состоит в отказе от потребления тех продуктов, которые в большом количестве содержат пуриновые основания. Исключаются пиво, кофе, чай, шоколад, мясные продукты, печень, красное вино. Предпочтение отдается вегетарианской диете с количеством чистой воды не менее двух литров в сутки.
Медикаментозный подход заключается в применении аллопуринола, конкурентного ингибитора ксантиноксидазы, — той самой, что переводит ксантины в мочевую кислоту. Это терапия первой линии [4–6].
В силу того, что аллопуринол отнимает все внимание ксантиноксидазы, превращаясь в хорошо растворимый аллоксантин, ксантин и гипоксантин, будучи обиженными на сложившуюся ситуацию, покидают организм с мочой [6].
Уратная нефропатия
Уратная нефропатия создает массу проблем. Надо сказать, что сам термин «уратная нефропатия» сегодня используется реже, чем ранее, а кем-то и вовсе считается неправильным. Несмотря на это, именно так мы назовем поражение почек при гиперурикемии. Часто именно ураты формируют мочевые камни, становясь основой для мочекаменной болезни и вторичного пиелонефрита. У примерно половины людей, страдающих подагрой, присутствует еще и мочекаменная болезнь [4].
Механизм камнеобразования в данном случае провоцируется повышением кислотности (понижением рН) мочи. Это делает возможным переход мочевой кислоты в плохо растворимую форму:
Мочекаменная болезнь опасна вследствие риска возникновения острой задержки мочи, которая становится причиной острой постренальной почечной недостаточности, а та, в свою очередь, является угрожающим жизни патологическим состоянием.
Синдром Леша — Нихана
В случае наличия мутации гена, кодирующего гипоксантингуанинфосфорибозилтрансферазу, развивается синдром Леша — Нихана. Наследуется он Х-сцепленно, а это значит, что достается практически только парням. Но не часто (реже, чем могло бы быть, но чаще, чем хотелось бы): патология грозит одному человеку из 380 тысяч.
Особенность этого синдрома в том, что здесь имеется грозная неврологическая и психическая симптоматика с самого раннего возраста: умственная отсталость и снижение темпов психомоторного развития; нарушения двигательных функций по типу хореатетоза, аутоагрессивное поведение (ребенок пытается нанести себе вред, что в условиях умственной отсталости закономерно; но по той же причине такие попытки не всегда удачны). К сожалению, патофизиологические механизмы развития поражения ЦНС при болезни Леша — Нихена не ясны по сей день. «Так, несмотря на широкий спектр проблем, связанных с гиперпродукцией мочевой кислоты, причинно-следственная связь между ними и неврологическими и поведенческими проявлениями не установлена, и терапия аллопуринолом, проводимая с момента рождения, не влияет на поражение ЦНС» [7].
Помимо поражения нервной системы такие дети не лишены и уратной нефропатии, а также подагры. Оба этих патологических состояния развиваются по рассмотренным нами выше механизмам [6, 7].
Последнее, что здесь стоит отметить: спектр клинических проявлений варьирует в зависимости от степени утраты активности фермента. Так, при активности фермента ниже 1,5 % от необходимой развивается полный набор вышеописанных клинических симптомов, в том числе и аутоагрессивное поведение, и когнитивная недостаточность. Это — следствие поражения коры головного мозга. При сохранении активности фермента в интервале от 1,5 до 8 % от необходимой признаки поражения коры, чаще всего, не прослеживаются.
Но имеются иные неврологические нарушения, проявляющиеся в разной степени выраженности экстрапирамидной и пирамидной моторной дисфункции и связанные с гиперурикемией. Сохранение активности фермента свыше 8 % сопровождается лишь уратной нефропатией (нефролитиазом — образованием камней в мочевыводящих путях) и подагрой [4, 6, 7].
Синдром лизиса опухоли
Ряд опухолевых заболеваний (чаще это гемобластозы — лейкозы, лимфомы, реже — солидные опухоли: рак желудка, легкого, молочной железы и т. д.) может давать картину острой почечной недостаточности в ответ на проводимую химиотерапию. Распадаясь и разрушаясь от действия химиотерапевтических препаратов (флударабина, метотрексата, да и многих других), опухолевые клетки выбрасывают в кровь большое количество мочевой кислоты, калия и фосфора. Последствия бывают достаточно серьезные:
- гиперурикемия провоцирует развитие нефролитиаза почечных канальцев и почечной недостаточности;
- гиперкалиемия способсвует возникновению тяжелых сердечных аритмий, вплоть до остановки сердца (усугубляется вторичной гипокальциемией) в связи с угнетением атриовентрикулярной проводимости в сердце;
- гиперлактатемия (связана с высокой активностью в опухолевых клетках анаэробного гликолиза, обусловливающего высокое содержание в них лактата) с риском развития метаболического ацидоза.
К метаболическим изменениям при синдроме лизиса опухоли также относят повышение уровня остаточного азота в крови (в частности, мочевины, которое выступает как проявление повышенного катаболизма белков клеток), вторичную гипокальциемию на фоне гиперфосфатемии, изменение кислотности крови — ацидоз.
Гиперурикемия при несоответствии возможностям почек провоцирует мочекислый нефролитиаз с развитием мочекислой нефропатии (по указанному в 1.6.2 механизму) и острой почечной недостаточности.
Тяжелый комбинированный иммунодефицит
Незаслуженно забываемый многими фермент аденозиндезаминаза может быть причиной тяжелого комбинированного иммунодефицита у детей раннего возраста. Заболевание развивается в случае мутации гена, кодирующего данный фермент. Аденозидезаминаза (АДА) превращает аденозин в инозин, тем самым участвуя в катаболизме адениловых нуклеотидов. При отсутствии АДА в клетках накапливается аденозин и дезоксиаденозин. Повышенное содержание этих метаболитов угнетает рибонуклеотидредуктазу — ту самую, что отвечает за синтез нуклеиновых кислот. Без них невозможен синтез ДНК, а значит, угнетается пролиферация клеток. Первыми отхватывают клетки крови (быстро пролиферирующие), в особенности лимфоциты, как В, так и Т. В результате развивается комбинированный иммунодефицит, т. е. дефект клеточного и гуморального адаптивного иммунитета. Все это проявляется чрезвычайно частым развитием гнойных тяжелых инфекций, которые для обычных детей не совсем характерны и приобретают рецидивирующий характер, не склонный к разрешению. Происходить это все начинает (приблизительно) с шести месяцев, когда заканчивается действие материнских антител, получаемых младенцем при грудном вскармливании. Дети болеют тяжело, часто, и причем такими инфекциями, которыми иммунокомпетентный ребенок болеть не будет (эти инфекции называют оппортунистическими) [2].
В настоящее время есть данные об успешной генотерапии (введении во взятые от пациента стволовые клетки костного мозга генетического вектора, кодирующего дезаминазу) с последующим возвращением и серией ТГСК (процедур трансплантации гемопоэтических стволовых клеток). Это два наиболее адекватных варианта лечения таких заболеваний, но, как ты понимаешь, они не везде доступны.
Не могу не отметить, что ТКИД лишь в 50 % случаев вызывается мутацией в гене, кодирующем дезаминазу. ТКИД как группа заболеваний обусловлен нарушением процесса созревания или пролиферации лимфоцитов. Существует 15 генетических вариантов данного заболевания. Иногда виноват дефект другого фермента катаболизма пуринов — пуриннуклеозидфосфорилазы, с которой мы встречались ранее.
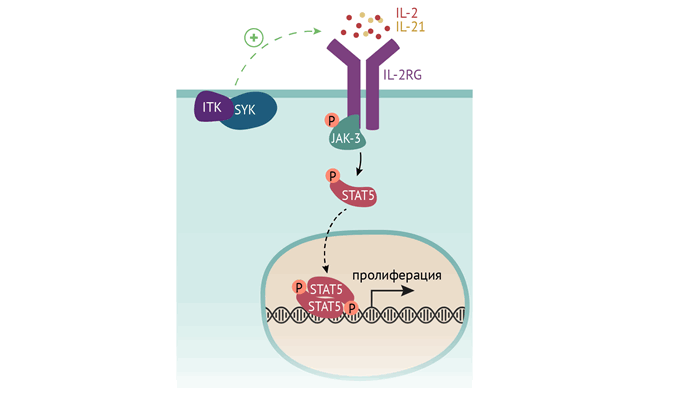
Также причина ТКИД может быть результатом дефекта IL2RG — общей субъединицы для многих рецепторов к цитокинам (ИЛ-2/4/7/9/15/21), запускающим клеточный ответ в лимфобластах в виде их пролиферации и созревания. Бывает изолированный дефект рецептора к ИЛ-7. А может быть дефект киназы JAK3 (янус-тирозинкиназы), которая является внутриклеточным посредником сигнала, получаемого вышеуказанными рецепторами.
Отдельной группой причин служит дефект рекомбиназ, осуществляющих реаранжировку генома лимфоцитов — того самого процесса, который обусловливает разнообразие спектра антител и Т-лимфоцитарных рецепторов к самым разным антигенам и необходимого для созревания В- и Т-лимфоцитов.
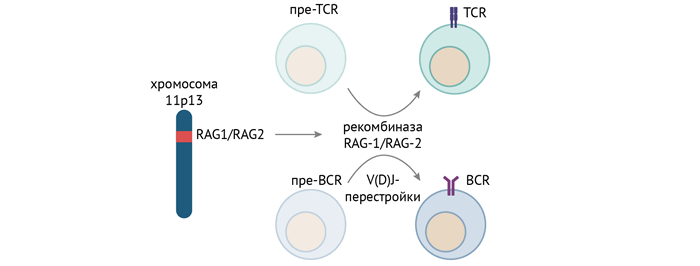
Несмотря на разнообразие причин, приводящих к ТКИД, клинический фенотип один: рецидивирующие и тяжелые инфекции, обусловленные нарушением созревания Т- и В-лимфоцитов [2].
Оротовая ацидурия I/ II типов
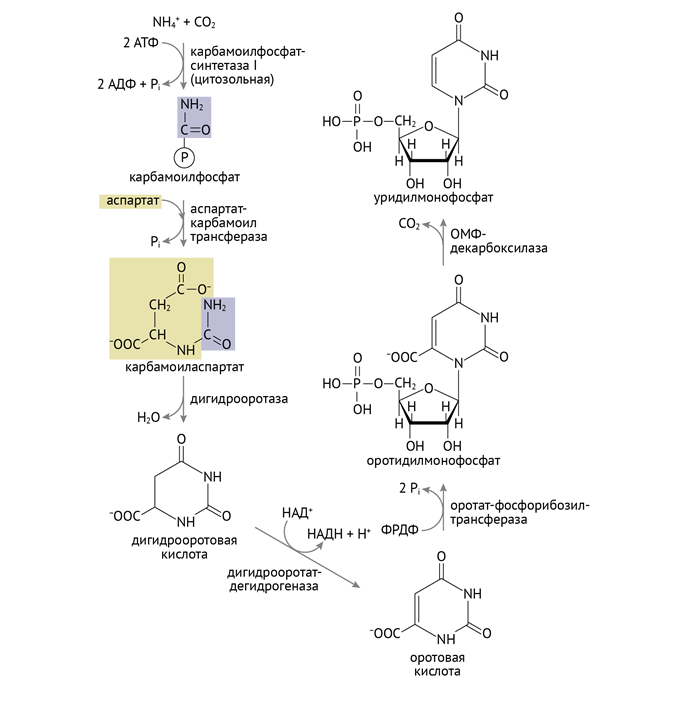
Нарушения пиримидинового обмена менее зрелищны. Одним из главных (и в то же время редких) таких нарушений пиримидинового обмена является оротовая ацидурия. Она обусловлена дисфункцией одного или двух ферментов:
- оротатфосфорибозилтрансферазы (превращает оротат в ОМФ);
- оротатфосфорибозилдекарбоксилазы (превращает ОМФ в УМФ) (изолированный дефект данного фермента вызывает II тип оротовой ацидурии, дефект обоих указанных ферментов — оротовую ацидурию I типа) .
Это ферменты конечных этапов синтеза УМФ — предшественника всех остальных пиримидинов. Нетрудно догадаться (если взглянуть на схему ниже и главу 1.2), что данная энзимопатия будет сопровождаться накоплением оротовой кислоты [1, 4, 6].
Специфическое клиническое проявление такой энзимопатии — оранжевая моча, которая характерна для первого типа и чье появление обусловлено непосредственным накоплением в ней оротовой кислоты. Но важно не это. А то, что этот дефект чреват нарушением синтеза пиримидинов, а значит, угнетением синтеза нуклеиновых кислот. Характерным будет развитие тяжелой мегалобластной анемии (анемии, обусловленной несовершенным гемопоэзом в результате нарушения репликации ДНК, когда клетка пытается поделиться надвое, а ДНК не хватает). Выглядит это примерно вот так:
Обычно такое характерно для нехватки витамина В12 и фолиевой кислоты — важных факторов, необходимых для репликации клеток. Их роль заключается в осуществлении превращения дезоксиуридилтрифосфата в дезокситиминтрифосфат — субстрат для ДНК-полимеразы и необходимый компонент для синтеза ДНК. В9 является коферментом тимидилатсинтетазы, а В12 — посредником восстановления активной формы В9 (см. главу 2). Дефицит этих витаминов — частая причина развития мегалобластной анемии. Логично предположить, что лечение такой анемии при оротовой ацидурии будут начинать с их введения, и это справедливо.
Однако эффекта будет чуть меньше, чем никакого. Ибо причина в том, что у нас в принципе нечего превращать в дТТФ. У нас нет уридина в клетках, и он не появится в силу наличия ферментного блока.
Зато разовьется мегалобластная анемия, иммунодефицит, ну и, конечно же, нефролитиаз (кристаллы оротовой кислоты также любят создавать камушки в почечных канальцах).
Выход есть. В таких случаях назначается пожизненное лечение рекомбинантным уридином. По «запасному» пути экзогенный уридин превратится в УМФ и далее по расписанию. Здесь важно вовремя заподозрить возможность наличия у ребенка такого дефекта.
Читайте также: