Приобретенный буллезный эпидермолиз
Добавил пользователь Владимир З. Обновлено: 27.01.2026
Над статьей доктора Захарцовой Инны Леонидовны работали литературный редактор Елизавета Цыганок , научный редактор Надежда Гавран и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Буллёзный эпидермолиз (Epidermolysis bullosa) — это болезнь, связанная с мутациями в разных генах. Степень тяжести и разнообразие клинических проявлений зависит от того, какой именно ген поражён.
Основной симптом — образование пузырей и эрозий на коже и слизистых оболочках, гиперчувствительность и ранимость кожи при незначительных механических травмах («механобуллёзная болезнь») [1].
Буллёзный эпидермолиз имеет две формы болезни: врождённую (ВБЭ) и приобретённую (ПБЭ) [3].
До 1886 года врождённый буллёзный эпидермолиз называли наследственной пузырчаткой. Впервые клиническую картину этой болезни описал британский доктор Дж. Хатчинсон в 1875 году [4][5][6][7].
При врождённом буллёзном эпидермолизе болезнь является результатом генной мутации, при которой дефектный ген передаётся от родителя к ребёнку [3]. Установлено 18 генов, которые могут стать причиной болезни. Если один из них даёт сбой, кожа становится настолько непрочной, что даже лёгкое давление, трение или химическое и температурное воздействие могут стать причинами образования нового пузыря [2].
Приобретённый буллёзный эпидермолиз — хроническое аутоимунное заболевание, для которого характерно образование пузырей под слизистыми оболочками и наружным слоем кожи (эпидермисом). В этом случае нарушаются межклеточные связи в самом эпидермисе или между ним и дермой, в которой расположены сосуды, потовые железы и волосяные луковицы.
Приобретённый буллёзный эпидермолиз проявляется хрупкостью кожи, пузырями, эрозиями и язвами на разных стадиях заживления кожи. Для него также характерно образование рубцов и белых угрей [3].
Распространённость буллёзного эпидермолиза
Статистический учёт больных буллёзным эпидермолизом в России отдельно не вёдется, поэтому точные данные о заболеваемости и распространённости неизвестны. Однако есть отдельные статистические данные, согласно которым средний показатель распространённости врождённого буллёзного эпидермолиза в России составляет 3,64 случая на 1 млн населения, а максимальный показатель распространённости приходится на Республику Дагестан — 19,73 случаев на 1 млн человек.
Высокий показатель распространённости (свыше 10 случаев на 1 млн населения) отмечают и в других регионах России, например в Томской области, Чеченской Республике, Республике Мордовии и Костромской области [3].
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы буллёзного эпидермолиза
Основным клиническим признаком любой формы буллёзного эпидермолиза является появление пузырей при незначительной механической травме, например порезе, натирании одеждой, ссадине, расчёсе и т. д. Если повредить эти пузыри, образуется открытая рана, в которую может проникнуть инфекция.
К другим клиническим признакам относят огрубение и ороговение поверхности ладоней и подошв, а также нарушение пигментации кожи: на ней могут появиться как яркие пятна, так и участки с потерей пигмента (депигментация). Эти нарушения возникают на месте лопнувших пузырей.
Скованность движений в суставах, сращение кожи между пальцами и отсутствие ногтей — частые проявления тяжело протекающих форм болезни.
Среди редко встречающихся признаков следует отметить выпадение волос, повышенное или недостаточное потоотделение, эрозии или пузыри в полости рта, кариес, сужение ротовой щели, затруднение глотания, рвоту, запор и диарею [5][8].
Даже незначительное увеличение температуры окружающей среды может ухудшить состояние кожи, провоцируя образование новых пузырей и/или эрозий.
Врождённая форма болезни проявляется с рождения или первых месяцев жизни и характеризуется непрерывным течением с периодическими обострениями.
Приобретённый буллёзный эпидермолиз у детей встречается чаще чем у взрослых. Клинические проявления этой формы напоминают течение буллёзного пемфигоида, когда к образованию пузырей присоединяется ещё и зуд с вовлечением слизистых оболочек. Пациенты с этой формой болезни обычно отмечают высокую эффективность монотерапии с использованием Дапсона [14].
Буллёзный эпидермолиз у взрослых часто сочетается с болезнями желудочно-кишечного тракта, например сужением просвета пищевода, анальными трещинами, запором, воспалительными болезнями толстого и тонкого кишечника, а также с системными аутоиммунными заболеваниями, сахарным диабетом и лимфомой [3].
Патогенез буллёзного эпидермолиза
Кожа человека состоит из эпидермиса, дермы и подкожно-жировой клетчатки. Эпидермис включается в себя пять различных слоёв эпителиальных клеток, или кератиноцитов, самый нижний из которых — базальный эпителий — прикрепляется к дерме с помощью множества разных белков. Эти белки не только определяют стабильность соединения клеток базального эпителия с дермой, но и их собственную прочность.
Врождённый буллёзный эпидермолиз является результатом мутации генов, которые кодируют структурные белки кожи. Снижение синтеза белков или полное его отсутствие приводит к разделению слоёв кожи и появлению пузырей. Уровень, на котором в коже формируется пузырь, и, соответственно, клиническая форма заболевания зависят от места, где находится дефектный белок [3].
Приобретённый буллёзный эпидермолиз появляется, когда организм сам разрушает коллаген VII типа. Этот тип коллагена отвечает за связь эпидермиса с дермой, в то время как сам коллаген — это белок, который обеспечивает прочность и эластичность соединительной ткани [12]. Повреждение этой структуры нарушает связь эпидермиса с дермой и провоцирует спонтанное возникновение пузырей [15]. Причина такого поведения организма до сих пор неизвестна.
Классификация и стадии развития буллёзного эпидермолиза
В соответствии с консенсусом Третьего международного согласительного совещания, принято выделять четыре основных типа врождённого буллёзного эпидермолиза:
Среди клинических форм приобретённого буллёзного эпидермолиза выделяют два типа:
Описаны случаи развития одновременно классической и воспалительной форм ПБЭ [17].
Также учёные выделяют третий тип болезни, у которого нет официального названия. Он похож на рубцующийся пемфигоид, когда в патологический процесс вовлечены преимущественно слизистые оболочки с последующим их рубцеванием [3].
Ещё одна форма, при которой пузыри формируются под эпидермисом, напоминает течение линейного IgA-зависимого буллёзного дерматоза. Для него характерно поражение кожи туловища, рук, ног, лица и волосистой части головы, а также слизистых оболочек рта и глаз. У детей встречается редко [3].
Осложнения буллёзного эпидермолиза
Осложнения буллёзного эпидермолиза делятся на кожные и внекожные.
Кожные осложнения характерны для врождённого буллёзного эпидермолиза. К ним относятся:
- Большие пигментные родинки (невусы) от светло-бежевого до тёмно-коричневого и чёрного цвета, которые с возрастом светлеют.
- Повышенный риск меланомы у детей, даже на внешне нормальных участках кожи.
- Агрессивный, часто первично-множественный плоскоклеточный рак, устойчивый к химио- и лучевой терапии.
- Псевдосиндактилия, которой присуще не только сращение пальцев рук и ног, но и их атрофия, ограниченность или полная утрата движений большого пальца, кистей и стоп. Есть случаи рецидивов, которые возникают после операции, направленной на восстановление двигательной активности суставов.
Внекожные осложнения, характерные для обеих форм болезни:
- Поражение полости рта и желудочно-кишечного тракта, например тяжёлый кариес, ранняя потеря зубов, нарушение созревания твёрдых тканей зубов и частичное или полное отсутствие эмали, сужение просвета пищевода и заднего прохода, хронический запор, болезненная дефекация. В некоторых случаях пища или жидкость может попасть в нос, гортань и трахею, а глотание приносит боль.
- Поражение дыхательных путей, например отёк слизистых, пузыри, эрозии, рубцевание, охриплость голоса, сужение гортани, острая непроходимость дыхательных путей, которая сопровождается затруднением дыхания, одышкой и др.
- Поражение органов зрения, например эрозии и рубцевание роговицы, сращение одного или обоих век с глазным яблоком из-за появления пузырей на веках и слизистых, недостаточная выработка слёзной жидкости, воспаление краёв век и ухудшение зрения.
- Поражение мочеполовой системы, например болезненное мочеиспускание, сужение и непроходимость мочевых путей, пузырно-мочеточниковый рефлюкс, гидронефроз, почечная гипертония, гломерулонефрит, амилоидоз, почечная недостаточность и уросепсис.
- Ухудшение метаболизма и общего состояния (при тяжёлых формах буллёзного эпидермолиза). Из-за обнажения больших участков дермы организм расходует больше калорий, чем получает, поэтому появляется дефицит питательных веществ и белков. Ребёнок начинает отставать в росте, плохо себя чувствует из-за развивающейся анемии, у него медленно заживают раны и часто возникают инфекционные заболевания [3].
Диагностика буллёзного эпидермолиза
Диагноз «врождённый буллёзный эпидермолиз» традиционно устанавливают на основании симптомов и истории болезни, например по возникновению пузырных высыпаний или тенденции к их формированию с рождения, в младенческом или раннем детском возрасте. Чаще они появляются после незначительных физических и химических воздействий или спонтанно. Но клинических признаков и анамнеза недостаточно для постановки правильного диагноза — необходимо провести дополнительное молекулярно-генетическое исследование, которое выявляет мутацию генов, вызывающих патологию.
Диагностика врождённого буллёзного эпидермолиза
Симптомы врождённого буллёзного эпидермолиза могут отличаться даже в случае одной мутации или, наоборот, быть похожими при мутациях разных генов. Поэтому для установления субтипа болезни необходимо изучить биопсийный материал с помощью:
- трансмиссионной электронной микроскопии кожи — определяет уровень локализации пузыря и другие ультраструктурные изменения;
- прямой и непрямой иммунофлуоресцентной микроскопии — помимо местонахождения пузыря выявляет молекулярный дефект, лежащий в основе заболевания, т. е. наличие, отсутствие или недостаточный синтез искомого белка;
- генетической диагностики — обнаруживает мутантный ген [16].
Эти исследования дают возможность уточнить диагноз врождённого буллёзного эпидермолиза и определить его клиническую форму [11]. Однако такие обследования можно провести не в любой клинике.
Диагностика приобретённого буллёзного эпидермолиза
С учётом неоднородности клинических проявлений диагностика приобретённого буллёзного эпидермолиза, основанная исключительно на внешних симптомах, затруднительна. В связи с этим применяют следующий алгоритм поиска:
Лечение буллёзного эпидермолиза
Пациентов с подозрением на буллёзный эпидермолиз на всех этапах лечения консультируют дерматолог и генетик.
Лечение врождённого буллёзного эпидермолиза
Лечение врождённого буллёзного эпидермолиза в первую очередь основано на симптоматических методах. Симптоматическое лечение предотвращает образование пузырей или снижает их количество с помощью различных защитных средств.
Основной целью ухода за ранами является заживление кожи и слизистых оболочек. Большинство ран больных буллёзным эпидермолизом покрывют специальными атравматичными неприлипающими материалами (специальными пластырями), а после накладывают несколько слоёв бинтов. В некоторых случаях рекомендуют хирургическую обработку ран, например при их инфицировании.
Для исправления деформаций кисти и стопы может потребоваться хирургическое вмешательство, например реконструктивно-пластическая операция или артродез (полное обездвиживание поражённых суставов).
Лечение приобретённого буллёзного эпидермолиза
Больных приобретённым буллёзным эпидермолизом лечат медикаментами, но чаще всего такая терапия неэффективна. Во многих случаях пациенты не реагируют на высокие дозы системных гормональных препаратов, например Азатиоприна, Метотрексата и Циклофосфамида.
Вместе с тем в ряде случаев показана эффективность монотерапии Дапсоном, а также его комбинации с Преднизолоном. Эти средства направлены на профилактику и лечение высыпаний и оказывают противовоспалительное, антибактериальное и противогрибковое действие.
Эффективен в лечении и Циклоспорин, однако токсичность препарата не позволяет широко его использовать [3].
Ещё одним методом является плазмаферез, при котором из плазмы удаляют токсичные вещества. Этот способ помогает добиться контроля над антителами к коллагену VII типа.
Отмечалась также эффективность использования внутривенного иммуноглобулина. В некоторых клинических исследованиях показаны положительные результаты применения:
- биологических препаратов против антигена фактора некроза опухоли-альфа (ФНОα);
- антител к CD-антигену против В-лимфоцитов, продуцирующих антитела [3].
Также всем пациентам показана поддерживающая системная терапия, при которой следует:
- ухаживать за открытыми эрозивно-язвенными поражениями кожи, например обрабатывать антисептическими растворами и растворами с анилиновыми красителями;
- избегать травм, в том числе защищать участки кожи, наиболее подверженные травмам, использовать одежду без грубых швов и резинок, соблюдать охранительный режим и др. [3]
Прогноз. Профилактика
Прогноз буллёзного эпидермолиза во многом зависит от подтипа болезни. Большинство пациентов с ВБЭ, особенно с простым и дистрофическим буллёзным эпидермолизом, могут с высокой вероятностью почти полностью избавиться от проявлений болезни и вести нормальную жизнь, хотя не исключена возможность возникновения серьёзных осложнений.
Напротив, для пациентов с пограничным буллёзным эпидермолизом прогноз неблагоприятный: большинство больных могут умереть в течение первых нескольких лет жизни, также они подвержены серьёзному риску осложнения в виде метастатического плоскоклеточного рака [18].
Пациенты с ПБЭ при правильном лечении и уходе имеют примерно такую же продолжительность жизни, как и здоровые люди. Прогноз одного ретроспективного анализа показал, что среднее время до ремиссии составляет 9 месяцев. Полная ремиссия через 1 год, 3 года и 6 лет наступила у 33, 33 и 45 % пациентов соответсвенно [19]. Но даже при полной ремиссии для контроля болезни требуется длительная поддерживающая терапия.
Приобретённый буллёзный эпидермолиз редко приводит к смерти, но болезнь также может вызывать серьёзные осложнения. Кроме того, возможны проявления побочного эффекта от лекарств, используемых для лечения ПБЭ [20].
Профилактика буллёзного эпидермолиза
Чтобы оценить риск рождения ребёнка с буллёзным эпидермолизом и развития наследственного заболевания, проводится пренатальная диагностика — медико-генетическое консультирование семьи. Показаниями для консультирования являются:
- рождение ребёнка с врождённым буллёзным эпидермолизом;
- наличие болезни у одного из родителей;
- установленное или предполагаемое заболевание в семье.
Генетический анализ образцов крови и кожи больного позволяет уточнить тип и субтип врождённого буллёзного эпидермолиза, а также обнаружить мутацию соответствующего гена. Это очень важно для определения методов диагностики и тактики лечения болезни.
При беременности на сроке 10–12 недель проводят биопсию ворсин хориона с дальнейшим молекулярным исследованием полученного материала. Результаты получают в течение 3–4 дней после взятия материала. Это позволяет быстро проконсультировать семью и определить тактику ведения беременности.
Пациентам с врождённым буллёзным эпидермолизом для профилактики появления пузырей нужно беречь кожу и слизистые оболочки от травм, например носить свободную одежду, соблюдать диету, использовать нелипкие впитывающие повязки, ухаживать за полостью рта и т. д.
Для профилактики развития осложнений необходимо диспансерное наблюдение пациентов, в которое входит:
- периодическая сдача анализов для обнаружения и контроля анемии;
- полный осмотр больных для раннего выявления злокачественных опухолей кожи;
- своевременное лечение зубов [16].
В России есть только один фонд, который помогает людям с таким заболеванием — «БЭЛА. Дети-бабочки». Его участники помогают улучшить медицинскую помощь и всесторонне поддерживают людей с врождённой формой болезни.
Приобретенный буллезный эпидермолиз у детей: серия клинических случаев
Обоснование. Приобретенный буллезный эпидермолиз (ПБЭ) — хроническое заболевание, сопровождаемое образованием субэпидермальных пузырей на коже и слизистых оболочках в результате аутоиммунной агрессии к коллагену VII типа. Диагностика ПБЭ у детей затруднена из-за схожести клинической картины с другими пузырными дерматозами детского возраста.
Описание клинического случая. Представлено описание трех клинических случаев ПБЭ у детей. Показано, что для постановки правильного диагноза необходимо оценить клинические данные и определить глубину залегания пузырей по данным гистологического исследования биоптата кожи. Отложение IgG относительно базальной мембраны дермы по данным реакции непрямой иммунофлюоресценции помогает установить окончательный диагноз и определить тактику ведения пациента. В лечении детей с ПБЭ применялся препарат дапсон, показавший себя эффективным и безопасным средством первого выбора при ведении таких случаев.
Заключение. Представлен алгоритм дифференциальной диагностики ПБЭ у детей с другими пузырными заболеваниями детского возраста. Описаны результаты успешной лекарственной терапии заболевания.
Ключевые слова
Об авторах
доктор медицинских наук, заведующий отделением дерматологии с группой лазерной хирургии;
профессор кафедры дерматовенерологии и косметологии ЦГМА;
профессор кафедры педиатрии и детской ревматологии Первого МГМУ им. И.М. Сеченова,
119991, Москва, Ломоносовский пр-т, д. 2, стр. 1
Раскрытие интересов: Получение исследовательских грантов от фармацевтических компаний Jansen, Eli Lilly, Novartis. Получение гонораров за научное консультирование от компаний Galderna, Pierre Fabre, Bayer, Leofarma, Pfizer, AbbVie, ООО «Зелдис-Фарма».
Национальный медицинский исследовательский центр здоровья детей
Россия
Москва
Раскрытие интересов: Получение гонораров за научное консультирование от компаний Eli Lilly, Novartis.
Национальный медицинский исследовательский центр здоровья детей
Россия
Москва
Раскрытие интересов: Получение гонораров за научное консультирование от компаний Eli Lilly, Novartis, Jansen.
Национальный медицинский исследовательский центр здоровья детей
Россия
Москва
Раскрытие интересов: Получение гонораров за научное консультирование от компаний Eli Lilly, Novartis.
Список литературы
1. Roenigk HH Jr, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Arch Dermatol. 1971;103(1):1–10.
2. Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5(5):397–399. doi: 10.1177/120347540100500505.
3. Woodley DT, Chen M. Epidermolysis bullosa acquisita. In: Wolff K, Goldsmith LA, Katz SI, et al., editors. Fitzpatrick’s dermatology in general medicine. 8th ed. New York: McGraw-Hill Medical, 2015. pp. 705–713.
4. Klebes M, Wutte N, Aberer E. Dapsone as second-line treatment for cutaneous lupus erythematosus? A retrospective analysis of 34 patients and a review of the literature. Dermatology. 2016; 232(1):91–96. doi: 10.1159/000441054.
5. Russo I, Ferrazzi A, Zanetti I, Alaibac M. Epidermolysis bullosa acquisita in a 17-year-old boy with Crohn’s disease. BMJ Case Rep. 2015;2015. pii: bcr2015210210. doi: 10.1136/bcr2015–210210.
6. Kasperkiewicz M, Orosz I, Abeck D, et al. Childhood epidermolysis bullosa acquisita with underlying coeliac disease. Acta Derm Venereol. 2015;95(8):1013–1014. doi: 10.2340/00015555-2110.
7. Elliott GT. Two cases of epidermolysis bullosa. J Cutan Genitourin Dis. 1895;13:10.
9. Park SB, Cho KH, Youn JL, et al. Epidermolysis bullosa asquisita in childhood — a case mimicking chronic bullous dermatosis of childhood. Clin Exp Dermatol. 1997;22(5):220–222.
10. Caux F, Kirtschig G, Lemarchand-Venencie F, et al. IgA-epidermolysis bullosa asquisita in a child resulting in blindness. Br J Dermatol. 1997;137(2):270–275.
11. Asfour L, Chong H, Mee J, et al. Epidermolysis bullosa acquisita (brunsting-perry pemphigoid variant) localized to the face and diagnosed with antigen identification using skin deficient in type vii collagen. Am J Dermatopathol. 2017;39(7):e90–e96. doi: 10.1097/DAD.0000000000000829.
12. Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37(3):220–230. doi: 10.1111/j.1346-8138.2009.00799.x.
13. Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30(1):60–69. doi: 10.1016/j.clindermatol.2011.03.011.
14. Mayuzumi M, Akiyama M, Nishie W, et al. Childhood epidermolysis bullosa acquisita with autoantibodies against the noncollagenous 1 and 2 domains of type VII collagen: case report and review of the literature. Br J Dermatol. 2006;155(5):1048–1052. doi: 10.1111/j.1365-2133.2006.07443.x.
15. Lawrence A. Schachner, Ronald C. Hansen. Pediatric dermatology. 4th ed. Elsevier, 2011. pp. 978–979.
16. Kridin K. Accessible diagnostic methods to differentiate between epidermolysis bullosa acquisita and other subepidermal autoimmune bullous diseases. Indian J Dermatol. 2018;63(5): 445–448. doi: 10.4103/ijd.IJD_75_18.
17. Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol. 2004;22(2):129–138. doi: 10.1016/j.clindermatol.2003.12.020.
18. Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100(1):28–34.
19. Schneider-Yin X, Harms J, Minder EI. Porphyria in Switzerland, 15 years’ experience. Swiss Med Wkly. 2009;139(13–14): 198–206. doi: smw-12496.
20. Callot-Mellot C, Bodemer C, Caux F, et al. Epidermolysis bullosa acquisita in childhood. Arch Dermatol. 1997;133(9):1122–1126.
21. Connolly SM, Sander HM. Treatment of epidermolysis bullosa acquisita with cyclosporine. J Am Acad Dermatol. 1987;16(4):890.
22. Cunningham BB, Kirchmann TT, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. 1996;34 (5 Pt 1):781–784.
23. Tayal U, Burton J, Dash C, et al. Subcutaneous immunoglobulin therapy for immunomodulation in a patient with severe epidermolysis bullosa acquisita. Clin Immunol. 2008;129(3):518–519. doi: 10.1016/j.clim.2008.08.003.
24. Самцов А.В., Белоусова И.Э. Буллезные дерматозы. — Спб.: ООО «Издательско-полиграфическая компания «КОСТА»; 2012. — 144 с.
25. Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30(1):60–69. doi: 10.1016/j.clindermatol.2011.03.011.
Приобретенный буллезный эпидермолиз
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
Филиал "Вешняковский" Московского научно-практического центра дерматовенерологии и косметологии Департамента здравоохранения Москвы
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
ФГУ "Федеральный научный центр трансплантологии и искусственных органов им. В.И. Шумакова" Минздрава России, Москва
Особенности течения и диагностики приобретенного буллезного эпидермолиза (клинико-иммунопатологическое наблюдение)
Журнал: Клиническая дерматология и венерология. 2013;11(2): 92‑97
Махнева Н.В., Нефедова Е.Д., Померанцев О.Н., Белецкая Л.В. Особенности течения и диагностики приобретенного буллезного эпидермолиза (клинико-иммунопатологическое наблюдение). Клиническая дерматология и венерология. 2013;11(2):92‑97.
Makhneva NV, Nefedova ED, Pomerantsev ON, Beletskaia LV. The course and diagnosis of acquired epidermolysis bullosa (a clinical and immunopathological observation). Klinicheskaya Dermatologiya i Venerologiya. 2013;11(2):92‑97. (In Russ.).
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
Представлен клинический случай приобретенного буллезного эпидермолиза смешанного генеза с участием IgG/IgA-аутоантител, имитирующего клиническую картину вульгарной пузырчатки, торпидно протекающего на фоне иммуносупрессивной терапии. Рассматривая вопросы этиопатогенеза, авторы подчеркивают важную роль иммунофлюоресценции в диагностике и дифференциальной диагностике аутоиммунных буллезных дерматозов.
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
Филиал "Вешняковский" Московского научно-практического центра дерматовенерологии и косметологии Департамента здравоохранения Москвы
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
ФГУ "Федеральный научный центр трансплантологии и искусственных органов им. В.И. Шумакова" Минздрава России, Москва
Приобретенный буллезный эпидермолиз (ПБЭ) — редкое аутоиммунное буллезное заболевание кожи и слизистых оболочек, встречающееся в 0,17—0,26% случаев на 1 млн человек в Северной и Западной Европе [1, 2]. Несмотря на то что не обнаружено преимуществ выявления этого дерматоза в зависимости от расовой и половой принадлежности, отмечено его широкое распространение среди корейской популяции [3]. В Московской области за 1992—2006 гг. диагностировано всего 9 случаев ПБЭ при клинико-лабораторном обследовании 267 больных, страдающих аутоиммунными буллезными дерматозами [4].
Причина возникновения ПБЭ до конца не известна. Однако наличие в сыворотке крови у больных ПБЭ аутоантител, направленных к компонентам базальной мембраны кожи человека и животных (к детерминантам коллагена VII типа), свидетельствует о том, что буллезный дерматоз имеет аутоиммунный механизм развития [5—8]. Антитела больных ПБЭ относятся в основном к IgG [9, 10].
Основным клиническим признаком ПБЭ является возникновение спонтанных или травмоиндуцированных пузырей в зрелом возрасте (в 40—50 лет) с отсутствием семейной предрасположенности и исключением других буллезных дерматозов [11—13]. Как правило, в патологический процесс вовлекаются кожа и слизистая оболочка. При классическом варианте ПБЭ основным клиническим отличительным признаком является первичное возникновение пузырей и эрозивных дефектов на месте травм без или с минимальным проявлением воспалительного процесса. Это связано с физической слабостью кожного покрова, особенно в выступающих местах и местах сдавления (локтевых и коленных сгибов, сводов стоп) с образованием рубцов, милиа и/или эритемы [13—15]. Возможно развитие ониходистрофии и рубцовой алопеции [16, 17].
При воспалительном варианте ПБЭ пузыри, как правило, возникают на воспаленном участке кожи без травматизации на открытых ее участках. Клиническая картина отличается полиморфизмом и напоминает буллезный или рубцующийся пемфигоид [14, 18, 19].
Мы наблюдали за пациентом с ПЭБ смешанного генеза с участием IgG/IgA-аутоантител, имитирующего клиническую картину вульгарной пузырчатки, торпидно протекающего на фоне иммуносупрессивной терапии.
Обследован больной Ш., 54 лет. Болен около 6 лет (с 2006 г.). Впервые появились буллезные элементы на слизистой оболочке полости рта, коже волосистой части головы и верхней трети спины. Начало заболевания пациент связывает с избыточной инсоляцией и нервным перенапряжением. Клинически диагностирована вульгарная пузырчатка.
В Городской клинической больнице №14 им. В.Г. Короленко проведено лечение преднизолоном в максимальной начальной дозе (100 мг/сут внутрь) с постепенным ее снижением. Больной выписан с клинически положительным эффектом (эпителизацией эрозивных дефектов и отсутствием свежих буллезных элементов при дозе преднизолона 35 мг/сут). В течение 3 мес после выписки доза преднизолона была снижена до поддерживающей (10 мг/сут внутрь), на фоне которой периодически возникали единичные пузыри на коже конечностей, которые эпителизировались после применения спрея оксикорт. Патологический процесс постепенно прогрессировал и в 2009 г. пациент с обострением процесса госпитализирован в кожную клинику МИА им. И.М. Сеченова для обследования и коррекции терапии. При обследовании данных, свидетельствующих за паранеопластический процесс, не выявлено. Иммуноморфологическая картина интактного участка кожи свидетельствовала в пользу буллезного пемфигоида. Обнаружена фиксация IgG, IgA и С3-компонента комплемента в зоне базальной мембраны эпидермиса. Однако клинический диагноз «вульгарная пузырчатка» не был снят. Проведено лечение: метипред 20 таблеток в сутки (80 мг) в течение 21 дня. В связи с торпидным течением и отсутствием положительной динамики от проводимой терапии назначена комбинированная терапия преднизолоном (80 мг/сут) и азатиоприном (150 мг/сут). Однако патологический процесс прогрессировал.
К лечению были добавлены дипроспан (2,0 мл внутримышечно 1 раз в 5 дней, №3) и плазмаферез (8 сеансов). На фоне указанной терапии достигнут клинически положительный эффект (отсутствие свежих высыпаний, полная эпителизация эрозий). Пациент выписан на дозе преднизолона 25 мг/сут внутрь с рекомендациями о ее постепенном снижении. С 2010 г. пациент находится на поддерживающей терапии преднизолона (10 мг/сут внутрь). В течение последующих 1,5 лет у пациента периодически возникают единичные буллезные элементы и эрозивные дефекты на коже конечностей в местах травмирования, которые эпителизировались на фоне топических глюкокортикостероидов. С декабря 2011 г. отмечено обострение патологического процесса. Больной госпитализирован с диагнозом «вульгарная пузырчатка, обострение».
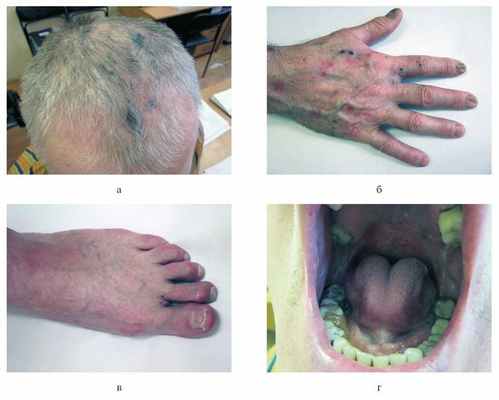
01.06.12 больной поступил в 1-е стационарное отделение филиала «Вешняковский» Московского научно-практического центра дерматовенерологии и косметологии Департамента здравоохранения Москвы (история болезни №12/246). Общее состояние удовлетворительное. Ведущие жалобы: высыпания на коже волосистой части головы, верхних и нижних конечностей, слизистой оболочке полости рта, жжение в местах высыпаний, боли при глотании и в эпигастрии. Пациент отмечал появление пузырей и эрозий непосредственно в местах ушиба или сдавления. При осмотре: процесс носит распространенный, подостровоспалительный характер с локализацией на коже волосистой части головы, туловища, верхних и нижних конечностей, слизистой оболочке полости рта. На коже волосистой части головы в проекции теменных зон процесс представлен эритематозно-сквамозными очагами и единичными эрозиями, расположенными на воспалительном фоне, покрытыми серозно-геморрагическими корочками размером до 1,5 см в диаметре (рис. 1, a). Рисунок 1. Клинические проявления ПБЭ. a — лобно-теменная зона: единичные эрозивные дефекты (до 0,5 см) на эритематозном фоне с серозно-геморрагическими корочками, эритематозносквамозные очаги; б — кожа правой кисти: эрозии (до 0,7 см в диаметре) с серозным отделяемым, серозно-геморрагическими корочками; поствоспалительные гиперпигментированные и эритематозные пятна (диаметром до 1,5 см), милиумы (до 0,3 см в диаметре); ногтевые пластины дистрофичны (тусклые, желтоватые, истонченные, продольно исчерченные); в — левая стопа: ногтевые пластины тусклые, деформированы, с продольной исчерченностью, множественными лейконихиями и крошащимися краями; г — полость рта. На коже туловища, конечностей — единичные эрозии до 0,7 см в диаметре, частично покрытые серозно-геморрагическими корочками с серозным отделяемым; на месте бывших высыпаний — гиперпигментированные пятна диаметром до 1,5 см. Симптом Никольского отрицательный. На коже кистей — милиумы (см. рис. 1, б). Ногтевые пластины тусклые, деформированы, продольно исчерчены, крошатся (см. рис. 1, в). На слизистой оболочке полости рта (щек, мягкого и твердого неба) патологический процесс представлен эрозиями с ярко-розовым дном размером до 0,7 см в диаметре, окаймленными по периферии обрывками эпителия беловатого цвета (см. рис. 1, г).
Из анамнеза: острые респираторные вирусные инфекции до 5—6 раз в год; в 2011 г. — операция по поводу правосторонней паховой грыжи.
Для исключения неопластического процесса как одного из факторов, провоцирующих буллезный дерматоз, пациенту проведено комплексное обследование. Диагностированы ишемическая болезнь сердца; стенокардия напряжения I—II функционального класса; гипертоническая болезнь II ст.; язвенная болезнь двенадцатиперстной кишки, рубцовая деформация луковицы двенадцатиперстной кишки; хронический панкреатит в ремиссии; мочекаменная болезнь, конкременты почек, киста синуса левой почки; хронический субатрофический фарингит. По данным эзофагогастродуоденоскопии, рубцовая деформация луковицы двенадцатиперстной кишки, поверхностный гастрит и косвенные признаки хронического панкреатита. В клиническом анализе крови — лейкоцитоз (до 15,3×10 9 /л). Данных за онкопатологию не обнаружено.
При цитологическом исследовании мазков-отпечатков со дна эрозий слизистой оболочки полости рта и кожи акантолитические клетки (клетки Тцанка) не обнаружены. При микроскопическом исследовании гладкой кожи и ногтевых пластин стоп грибы не выявлены.
На основании анамнеза, клинико-морфологической картины, особенности течения болезни и результатов ранее проведенного иммуногистохимического исследования пациенту выставлен диагноз ПБЭ.
Проведено лечение преднизолоном (60 мг/сут внутрь) с постепенным снижением дозы (до 50 мг/сут), на фоне которого на слизистой оболочке полости рта сохранялись эрозивные дефекты. Пациент переведен на комбинированную терапию глюкокортикостероидами: преднизолон (30 мг/сут) и метипред (16 мг/сут) внутрь. Лечение без эффекта. К терапии присоединен сандиммун-неорал в дозе 400 мг/сут ежедневно с последующим ее снижением до 300 мг/сут. На фоне приема сандиммун-неорала появились боли в эпигастральной и поясничной областях, и препарат был постепенно отменен. Комбинированный прием преднизолона (30 мг/сут внутрь) и метипреда (16 мг/сут внутрь) продолжен.
Из сопутствующей терапии: гемодез (200,0 мл внутривенно капельно через сутки, №3), трентал (5,0 мл внутривенно капельно с 200 мл физиологического раствора, №10), актовегин (5,0 мл внутримышечно ежедневно, №10), эссенциале (5,0 мл внутривенно струйно, №15), витамины В1 и В6 (по 1,0 мл внутримышечно, чередование), фолиевая кислота 1 мг (по 1 таблетке 2 раза в сутки), аспаркам (по 1 таблетке 3 раза в сутки), форкан 150 мг (1 капсула в неделю), омепразол 20 мг (1 капсула 2 раза в сутки), фосфалюгель (1 пакетик 3 раза в сутки), кальций D3-никомед 0,5 мг (по 1 таблетке 2 раза в сутки), эналаприл (10 мг 2 раза в сутки), индапамид (1,5 мг утром), гипотиазид (25 мг утром), нитроглицерин (0,5 мг при болях в области сердца). Местная терапия: на эрозивные дефекты на коже волосистой части головы, туловища — водный раствор метиленового синего, спрей оксикорт. Обработка слизистой оболочки полости рта тетраборатом натрия, полоскание полости рта отварами трав (шалфей, ромашка).
На фоне терапии эрозии на коже туловища, конечностей эпителизировались, на месте бывших высыпаний остались поствоспалительные пятна розово-красного цвета, милиумы на коже кистей, предплечий. На слизистой оболочке полости рта — эрозии в стадии эпителизации. Боли при глотании значительно уменьшились. Больной выписан с положительным клиническим эффектом на комбинированной терапии преднизолоном (30 мг/сут внутрь) и метипредом (16 мг/сут внутрь) под наблюдение дерматолога по месту жительства. Даны рекомендации.
ПБЭ — крайне редкая патология. Впервые ПБЭ описан во II половине XX века и получил свое название в связи с клиническим сходством с дистрофической формой врожденного буллезного эпидермолиза [20], который развивается у детей вследствие врожденного дефекта в гене, кодирующем коллаген VII типа (якорные фибриллы). Коллагены (фибриллярные и нефибриллярные) являются структурными элементами большинства форм соединительной ткани в организме человека и мутации в генах, кодирующих их, являются причиной развития разных заболеваний человека [21—25]. Это могут быть несовершенный остеогенез, некоторые формы остеопороза, хондродисплазия, сосудистые аневризмы, заболевание почек и буллезный эпидермолиз [26, 27]. В последнем случае дефектное связывание волокон коллагена VII типа с недостаточным количеством якорных фибрилл вызывает несостоятельность кожного покрова — его неустойчивость к механическим воздействиям с развитием травмоиндуцированных пузырей, которые могут располагаться как на спокойном, так и на минимально воспаленном фоне с образованием рубцов и милиа [13, 14, 16, 17]. Возможно развитие и ониходистрофии [11, 16, 17], что подтверждает и представленный нами клинический случай.
Ультраструктурное изучение кожи при ПБЭ позволило обнаружить аморфное состояние электронно-плотной полосы непосредственно под lamina densa с недостаточным количеством якорных фибрилл, которые крепят эпидермис к дерме, пересекая базальную мембрану [28—30]. Однако выявление в сыворотке крови у пациентов с данной патологией IgG-аутоантител к коллагену VII типа позволило предположить, что ПБЭ имеет аутоиммунный механизм развития, в котором якорные фибриллы скорее скомпрометированы аутоантителами, чем дефектами гена [6, 31]. Антитела не являются специфичными только для антигенов базальной мембраны эпидермиса. Они также взаимодействуют с антигенами базальных мембран слизистой оболочки полости рта, пищевода, влагалища [6, 12]. Ответственность аутоантител за проявления ПБЭ подтверждена экспериментально in vivo на новорожденных мышах [31—34]. Проведена пассивная передача аутоантител (очищенный IgG) от пациентов с ПБЭ новорожденным мышам. При этом у животных развивался процесс с гистологическими и иммунопатологическими изменениями, характерными для этого заболевания.
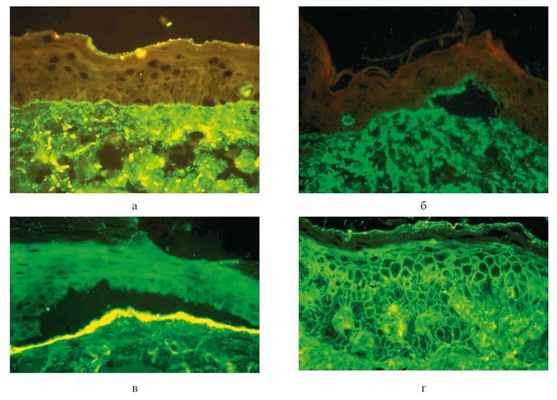
По мнению ряда авторов [35—37], в некоторых случаях патогенетическая роль при ПБЭ принадлежит циркулирующим аутоантителам класса A или одновременно IgA и IgG. Последнее подтверждает и описанный нами случай. С помощью методов меченых антител (прямой метод иммунофлюоресценции) в зоне базальной мембраны эпидермиса обнаружены связанные с тканями иммуноглобулины (IgA, IgG) и С3-компонент комплемента. Базальная мембрана эпидермиса является антигеном-мишенью для аутоантител буллезного пемфигоида и приобретенного буллезного эпидермолиза (рис. 2, a). Рисунок 2. Криостатные срезы кожи больных аутоиммунными буллезными дерматозами (для сравнительного контроля). Участки клинически непораженной кожи. Обработка меченой сывороткой против Ig G человека. Прямой метод иммунофлюоресценции (×400). a — буллезный пемфигоид: фиксация IgG в зоне базальной мембраны эпидермиса; б — в месте образовании пузыря фиксированный иммуноглобулин выявляется на покрышке в связи с фиксацией его в lamina lucida; в — фиксация IgG на дне подэпидермального пузыря (широкая щель) при ПБЭ; г — фиксация IgG в межклеточной субстанции всех слоев эпидермиса и на поверхности кожи при аутоиммунной пузырчатке. При этом фиксация аутоантител в lamina lucida и сохранение фиксированного иммуноглобулина на покрышке пузыря свидетельствуют о наличии буллезного пемфигоида (см. рис. 2, б), в то время как при ПБЭ фиксированный иммуноглобулин сохраняется на дне пузыря (см. рис. 2, в). При ПБЭ мишенью антител является lamina densa [38, 39].
В описанном нами случае ранее использованный метод меченых антител позволил исключить у пациента наличие аутоиммунной пузырчатки, для которой характерна картина фиксации IgG в межклеточной субстанции многослойного плоского эпителия (см. рис. 2, г) и заподозрить аутоиммунный буллезный дерматоз дермо-эпидермального соединения, в частности буллезный пемфигоид. Отсутствие подэпидермального микропузыря в криостатных срезах клинически интактного участка кожи пациента, к сожалению, не позволило иммуногистохимически уточнить мишень для фиксированных иммунных комплексов. Тем не менее тщательно собранный анамнез с указанием пациента на легкую ранимость кожного покрова, клинические проявления (рубцы, милиум, ониходистрофия), волнообразное течение болезни и торпидность патологического процесса к иммуносупрессивной терапии свидетельствовали в пользу ПБЭ.
Таким образом, ПБЭ — редкий буллезный дерматоз, имеющий специфические и неспецифические клинические проявления, лечение которого для врача представляет сложности. Это связано с волнообразным течением данного дерматоза, торпидного к иммуносупрессивной терапии. Неуклонное прогрессирование патологического процесса с вовлечением слизистых оболочек, угрожающего жизни пациента, требует от специалистов постановки точного диагноза с использованием молекулярно-биологических методов исследования, а от исследователей — изучения этиопатогенетических механизмов болезни с целью разработки более совершенных методов диагностики и лечения.
Научная электронная библиотека

Приобретенный буллезный эпидермолиз впервые описан во второй половине прошлого столетия и получил свое название в связи с клиническим сходством с дистрофической формой врожденного буллезного эпидермолиза. Последний развивается у детей вследствие врожденного дефекта в гене, кодирующем коллаген VII типа (якорные фибриллы), и проявляется неустойчивостью кожного покрова к механическим воздействиям с развитием травмо-индуцированных пузырей с образованием рубцов и милиум.
В основе диагностики приобретенного буллезного эпидермолиза лежит возникновение спонтанных или травмо-индуцированных пузырей в зрелом возрасте больного с отсутствием семейной предрасположенности и исключением других буллезных дерматозов. Различают два варианта приобретенного буллезного эпидермолиза: механобуллезный (классический или невоспалительный) и пемфигоидо-подобный (воспалительный).
При классическом варианте приобретенного буллезного эпидермолиза главным клиническим отличительным признаком является первичное возникновение пузырей и эрозивных дефектов на месте травм без воспалительного процесса или с минимальным его проявлением. Это связано с хрупкостью кожного покрова, особенно в выступающих местах и местах сдавления (локтевые и коленные сгибы, своды стоп). На месте эпителизации эрозивных дефектов формируются рубцовая атрофия, милиумы и/или эритема. Кроме того, у больных классическим приобретенным буллезным эпидермолизом патологический процесс первоначально может возникнуть на слизистой оболочке полости рта (слизистая щек, задняя стенка глотки, боковая поверхность языка, десна) и/или красной каймы нижней губы. Вовлечение в патологический процесс слизистых оболочек (полости рта, носа, пищевода, конъюнктивы, ануса, влагалища) может напоминать картину рубцующегося пемфигоида. Возможно развитие и стеноза пищевода как угрожающее жизни осложнение данного варианта приобретенного буллезного эпидермолиза. На рис. 15 представлены клинические признаки классического варианта приобретенного буллезного эпидермолиза.
При воспалительном варианте приобретенного буллезного эпидермолиза пузыри возникают на воспаленном участке кожи без травматизации (на открытых участках кожи тела). Кроме пузырей наблюдаются везикулы, склонные к слиянию с образованием гирляндоподобных элементов на эритематозном фоне, уртикарные и папулезные высыпания с вовлечением в патологический процесс слизистой оболочки полости рта. В ряде случаев поражается слизистая оболочка носа в виде геморрагических корочек и конъюнктивы (конъюнктивит). Картина может напоминать буллезный пемфигоид, реже герпетиформный дерматит Дюринга. На рис. 16 представлены клинические признаки воспалительного варианта приобретенного буллезного эпидермолиза.
В случаях приобретенного буллезного эпидермолиза паранеопластического генеза клиническая картина может быть представлена пузырями и обширными эрозивными дефектами на коже туловища и конечностей (рис. 17) с вовлечением в патологический процесс слизистой оболочки полости рта. Картина может напоминать вульгарную пузырчатку с положительным симптомом Никольского.
Одним из основных патогенетических факторов, способствующих развитию приобретенного буллезного эпидермолиза, признают наличие аутоантител к одному из компонентов базальной мембраны кожи человека и животных. Антитела не являются специфичными только для антигенов базальной мембраны эпидермиса, они также взаимодействуют с антигенами базальных мембран слизистой оболочки полости рта, пищевода, влагалища. Однако указанные антитела не реагируют с антигенами базальных мембран кровяносных сосудов, печени и эпителия паренхимы почки.
Локализация антигенов, к которым направлены антитела, совпадает с локализацией первичных деструктивных изменений. Образование подэпидермального пузыря (рис. 18, a) при приобретенном буллезном эпидермолизе начинается с расщепления дермо-эпидермального соединения под lamina densa или в верхних отделах дермы под комплексом lamina densa-якорные фибриллы (электронно-микроскопические данные). Установлено, что антигеном-мишенью для аутоантител является коллаген VII типа в пределах якорных фибрилл, которые проходят перпендикулярно от lamina densa к сосочковому слою дермы.
Ранее признанным аутоантигеном приобретенного буллезного эпидермолиза считалась α-цепь молекулярной массой 290 кД коллагена VII типа. Дальнейшее изучение строения коллагена данного типа на молекулярно-биологическом уровне позволило выявить, что антигеном-мишенью для этого буллезного дерматоза является ряд эпитопов молекул коллагена. А именно NC1 домен содержит главные иммунодоминирующие антигенные эпитопы для аутоантител приобретенного буллезного эпидермолиза. Этот домен может способствовать соединению коллагена VII типа с другими компонентами базальной мембраны и матрикса. Возможно, что аутоантитела, направленные против NC1, нарушают функцию этих адгезивных протеинов так, что якорные фибриллы коллагена полноценно не взаимодействуют с ними, а связываются с другими соединительнотканными компонентами зоны базальной мембраны и сосочкового слоя дермы. Такое изменение, т.е. потеря функции якорных фибрилл, ведет к нарушению дермо-эпидермального соединения.
В последующем было продемонстрировано, что некоторые аутоантитела у больных, страдающих приобретенным буллезным эпидермолизом, могут иметь мишенью не только NC1 домен, но и NC2 домен со спиральным (центральным) коллагеновым доменом. Это позволяет предположить, что последние также могут играть роль в поддержании функциональной полноценности коллагена VII типа и якорных фибрилл.
Ряд авторов считает, что большинство аутоантител в сыворотке крови пациентов относятся как к комплемент-активируемым, так и комплемент-неактивируемым субклассам IgG. При этом факт присутствия или отсутствия комплемент-активирующих аутоантител субклассов IgG в крови, титр которых может достигать 1:640, не коррелирует с клиническими формами заболевания. Это позволяет предположить, что аутоантитела могут иметь разную природу, в равной степени способствующие формированию пузырей как при комплемент-зависимом воспалительном повреждении, так и комплемент-независимом процессе, сопровождающимся механическим разрывом якорных фибрилл коллагена VII типа. Участие системы комплемента приводит к более выраженным клиническим проявлениям воспалительного приобретенного буллезного эпидермолиза, который клинически сходен с буллезным пемфигоидом и рубцующимся пемфигоидом.
По мнению ряда авторов в некоторых случаях патогенетическая роль при указанной патологии принадлежит циркулирующим аутоантителам класса IgA или одновременно IgA и IgG. С помощью непрямого метода иммунофлюресценции в сыворотке крови больных выявляют антитела IgG и/или IgA к антигенам базальной мембраны эпидермиса, а именно к антигенам lamina densa (рис. 18, b).
В ходе иммуноморфологического исследования биоптатов кожи больных приобретенным буллезным эпидермолизом первоначально были выявлены отложения IgG в зоне базальной мембраны эпидермиса, сходные с таковыми при буллезном пемфигоиде. В дальнейшем было обнаружено, что при буллезном пемфигоиде депозиты IgG располагаются в пределах полудесмосом и lamina lucida, а при приобретенном буллезном эпидермолизе - ниже, в пределах lamina densa и sublamina densa (иммуноэлектронная микроскопия). Прямым методом иммунофлюоресценции в криостатных срезах клинически непораженных участков кожи больных приобретенным буллезным эпидермолизом отмечается выраженная фиксация IgG в виде линейных отложений в зоне базальной мембраны эпидермиса, а в месте формирования пузыря - на его дне (lamina densa), что является диагностическим признаком данного буллезного дерматоза (рис. 18, c). В той же локализации могут быть обнаружены IgA, IgM, С3-компонент комплемента, но в меньшей степени интенсивности реакции.
Итак, приобретенный буллезный эпидермолиз - редкое аутоиммунное заболевание кожи и слизистых оболочек, который помимо классического течения может протекать атипично и имитировать другие буллезные дерматозы, такие как буллезный пемфигоид, рубцующийся пемфигоид или линейный IgA-зависимый дерматоз. В связи с этим имеются сложности в постановке клинического диагноза. Дифференцировать этот буллезный дерматоз позволяют только результаты иммунологических и иммуноморфологических исследований.
Рисунок 15. Больная приобретенным буллезным эпидермолизом (классический вариант).
a - кожа предплечья: ближе к области локтевого сустава располагается пузырь с прозрачным содержимым, плотной покрышкой на слегка гиперемированном фоне;
b - кожа нижних конечностей: около коленных суставов на месте травм располагаются эрозивные дефекты с ярко-красным дном, в области коленного сустава - множественные милиумы, расположенные группами на слегка гиперемированном фоне (справа);
c - слизистая языка: на боковой поверхности языка симметрично располагаются спавшиеся пузыри с плотной покрышкой белесоватого цвета, на кончике языка - пузырек размером до 0,5 см в диаметре с плотной покрышкой и серозным содержимым;
d - кожа кистей: на тыльной поверхности кисти располагаются в местах травм эрозивные дефекты, ногтевые пластины утолщены, отслаиваются.
Рисунок 16. Приобретенный буллезный эпидермолиз (воспалительный вариант).
a, b - кожа плечевого пояса: везикулезно-буллезные элементы, расположенные на слегка или сильно гиперемированном фоне, имеющие тенденцию к группировке по типу «нанизанных бус» (a) или рассеянно разбросанных пузырных элементов (b);
c - кожа предплечья: буллы с твердой покрышкой (частично спавшиеся) и прозрачным содержимым, расположенные на гиперемированном фоне;
d - кожа бедра: пузырь с твердой покрышкой и прозрачным содержимым, расположенный на слегка гиперемированном фоне, множественные эрозивные дефекты различных размеров на месте вскрывшихся пузырей.
Рисунок 17. Больная приобретенным буллезным эпидермолизом паранеопластического генеза.
a - тотальное поражение кожи с образованием обширных эрозивных поверхностей на месте пузырей, заживление которых происходит с формированием стойкой гиперпигментации;
b - на месте заживления эрозивного дефекта (гиперпигментация), по периферии образуются свежие «кольцевидные» буллезные элементы с твердой покрышкой и прозрачным содержимым.
Рисунок 18. Материалы больных приобретенным буллезным эпидермолизом. х 400.
a - криостатный срез биоптата кожи больной приобретенным буллезным эпидермолизом. Участок клинически интактной кожи. Окраска гематоксилином и эозином. Начальная стадия формирования подэпидермального пузыря - щель между эпидермисом и дермой (стрелка справа) и подэпидермальный пузырь (стрелка слева);
b - криостатный срез кожи донора здорового человека, обработанный сывороткой больной приобретенным буллезным эпидермолизом. Непрямой метод иммунофлюоресценции. Фиксация IgG в зоне базальной мембраны эпидермиса, в месте обычного для заболевания расслоения эпидермиса от дермы;
c - криостатный срез кожи больного приобретенным буллезным эпидермолизом. Прямой метод иммунофлюоресценции. Характерная фиксация IgG на дне (стрелка) подэпидермального пузыря (lamina densa).
Читайте также: