Синдром Розенберга—Хаториана: атрофия зрительных нервов, полинейропатия и глухота
Добавил пользователь Morpheus Обновлено: 27.01.2026
Синдром Розенберга-Чуториана - чрезвычайно редкое генетическое заболевание, характеризующееся триадой потери слуха, дегенерации зрительного нерва (атрофия зрительного нерва) и неврологическими нарушениями, в частности заболеванием нервов вне центральной нервной системы (периферическая нейропатия). Руки и ноги чаще всего поражаются периферической невропатией.
Признаки и симптомы
Симптомы синдрома Розенберга-Чатториана часто проявляются во время младенчества или раннего детства. Клиническая триада потери слуха, атрофии зрительного нерва и периферической невропатии характеризует расстройство.
У людей с синдромом Розенберга-Чуториана развивается сенсоневральная потеря слуха. У людей с типом нарушения слуха звук может проводиться обычно через наружное и среднее ухо. Однако звуковые колебания не передаются должным образом в мозг из-за дефекта внутреннего уха или слухового нерва, что приводит к потере слуха. (При нормальном слухе часть внутреннего уха служит для преобразования звуковых колебаний в нервные импульсы, которые затем передаются через слуховой нерв в мозг.) Хотя такая сенсоневральная потеря слуха обычно присутствует при рождении.
У индивидуумов с синдромом Розенберга-Чатториана также развивается дегенерация (атрофия) зрительного нерва. Оптический нерв - это структура, которая посылает электрические импульсы от сетчатки к мозгу. Оптическая атрофия приводит к потере остроты зрения.
Невоспалительным заболеванием, поражающим многие нервы (полинейропатии), является еще один признак синдрома Розенберга-Чуториана. Нарушаются нервы за пределами центральной нервной системы, особенно на руках и ногах (периферическая невропатия). У людей может развиться слабость и истощение (атрофия) мышц рук и ног. Разрушение жировых оболочек, окружающих нервы (демиелинизация), имело место у нескольких пораженных лиц.
Причины
Синдром Розенберга-Чуториана вызван мутацией в гене фосфорибозилпирофосфатсинтетазы I (PRPS1), расположенном на Х-хромосоме.
Синдром Розенберга-Chutorian наследуется как X-связанное нарушение. X-связанные генетические нарушения - это состояния, вызванные аномальным геном на Х-хромосоме и встречающиеся в основном у мужчин. Женщины, у которых есть ген болезни, присутствующий на одной из их Х-хромосом, являются носителями этого расстройства. Женщины-перевозчики обычно не проявляют симптомов, потому что у женщин есть две Х-хромосомы, и одна инактивируется, так что гены на этой хромосоме не функционируют. Обычно инактивируется Х-хромосома с ненормальным геном. Однако у некоторых женщин, несущих мутацию гена PRPS1, наблюдаются различные симптомы. У мужчин есть одна Х-хромосома, которая унаследована от их матери, и если самец наследует Х-хромосому, которая содержит ген болезни, он развивает болезнь. Женщины-носители X-сцепленного расстройства имеют 25% -ный шанс с каждой беременностью иметь дочерей-носителей, подобных им, 25% -ную вероятность иметь дочь-не-носителя, 25% -ную вероятность иметь сына, страдающего этим заболеванием, и 25% шанс получить незатронутого сына.
Мужчины с Х-сцепленным расстройством передают ген болезни всем своим дочерям, которые будут носителями. Мужчина не может передать ген X-сцепления своим сыновьям, потому что самцы всегда передают свою Y-хромосому вместо своей Х-хромосомы потомкам-мужчинам.
Затронутые группы населения
Синдром Розенберга-Chutorian - редкое генетическое нарушение, которое затрагивает мужчин чаще, чем женщин. Симптомы являются более серьезными у мужчин. Некоторые женщины-носители могут проявлять симптомы расстройства.
Связанные расстройства
Симптомы следующих расстройств могут быть сходными с симптомами синдрома Розенберга-Чуториана. Сравнения могут быть полезны для дифференциального диагноза:
Болезнь Шарко-Мари-Мари включает в себя группу наследственных невропатий, в которых поражаются моторные и сенсорные периферические нервы, что приводит к мышечной слабости и атрофии, главным образом в ногах, а иногда и в руках. Наследственная нейропатия CMT поражает нервы, которые контролируют многие мышцы тела. Нервные клетки у людей с этим расстройством не могут правильно посылать электрические сигналы из-за аномалий в аксоне нерва или аномалий изоляции (миелина) вокруг аксона. Специфические генные мутации ответственны за аномальную функцию периферических нервов. Charcot Marie Tooth наследственная невропатия может наследоваться в аутосомно-доминантном аутосомно-рецессивном или X-связанном режиме наследования.
Диагностика
Диагноз синдрома Розенберга-Чуториана основан на тщательной клинической оценке, подробной истории болезни и идентификации характерных результатов. Молекулярно-генетическое тестирование мутаций генов PRSP1 доступно для подтверждения диагноза. Тестирование носителей и пренатальная диагностика доступны, если мутация гена PRSP1 была идентифицирована у пострадавшего члена семьи.
Стандартные методы лечения
Лечение синдрома Розенберга-Чатториана направлено на конкретные симптомы, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий группы специалистов. Педиатры, неврологи, патологоанатомы, специалисты, которые оспаривают и лечат слуховые проблемы (аудиологи), глазные специалисты и другие медицинские работники, возможно, должны систематически и всесторонне планировать лечение пострадавшего ребенка.
Физическая и трудовая терапия может быть полезна для поддержания максимально возможной функциональности. Кохлеарный имплантат может помочь людям с потерей слуха. Подтяжки и другие ортопедические приспособления также могут помочь при ходьбе и движении.
Генетическое консультирование рекомендуется для пострадавших лиц и их семей.
Синдром Розенберга—Хаториана: атрофия зрительных нервов, полинейропатия и глухота
Синдром Розенберга—Хаториана: атрофия зрительных нервов, полинейропатия и глухота
Синдром, хараткеризующийся полинейропатией, атрофией зрительных нервов и нейросенсорной глухотой описали Rosenberg и Chutorian у 2 братьев и их племянника. Iwashita с сотр. сообщили о сибсах, имевших сходные аномалии. Менее подробно документированный случай был представлен Taylor. У сибсов, описанных Jequier и Deonna, диагноз обсуждается.
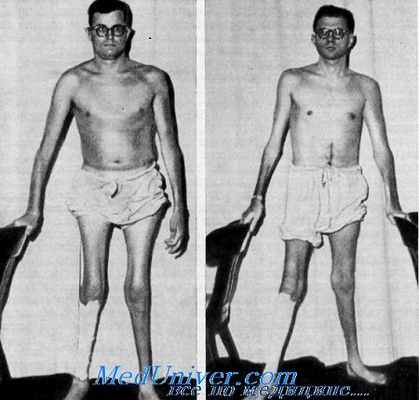
Клинические данные. Данные осмотра. Больные были нормального роста и телосложения, кроме мышечных атрофии, охватывающих конечности.
Орган зрения. У больных, о которых сообщили Rosenberg и Chutorian, потеря зрения была впервые отмечена (у обоих старших больных) в возрасте около 20 лет. Нарушения начались с никталопии, которая постепенно привела к снижению остроты зрения в дневное время. Офтальмологическое исследование братьев в возрасте 29 и 32 лет выявило атрофию зрительных нервов, выраженную резче в височных отделах, по не обнаружило пигментного ретинита. Корригированная острота зрения у одного брата с обеих сторон была 20/100, у другого — 20/400. Замечались только размашистые движения рук. Исследование полей зрения обнаружило у одного брата двустороннее концентрическое сужение. При обследовании их племянника в 3-летнем возрасте у него было выявлено нормальное зрение, по обнаружена атрофия зрительных нервов.
У больных, о которых сообщили Iwashita с сотр., в противоположность тому, что наблюдалось у больных Rosenberg и Chutorian, мышечные атрофии, потеря зрения и слуха развились в возрасте около 5 лет.
Нервная система. У больных, описанных Rosenberg и Chutorian, развитие моторики было задержанным, они начинали ходить почти в 2-летнем возрасте. Между 5 и 10 годами походка нарушилась в связи с атрофиями мышц ног; в конце концов больные были вынуждены опираться на палку. Интеллект был нормальным. Неврологическое исследование взрослых выявило резкую слабость и атрофию мышц дистальных отделов всех конечностей, включая собственные мышцы кистей и стоп. Мышцы лица, шеи и туловища были пощажены. Отмечалось снижение или отсутствие глубоких сухожильных рефлексов на ногах. Была также снижена тактильная и проприоцептивная чувствительность в дистальных отделах конечностей и обнаружено снижение скорости проведения нервных импульсов с отчетливой дснервацией.
У 3-летнего мальчика были выявлены некоторые нарушения походки и отсутствие глубоких сухожильных рефлексов. Мышечная сила и чувствительность были нормальными. У больных, описанных Iwashita с сотр., отмечались нарушения походки, координации, снижение чувствительности и положительная проба Ромберга.
Орган слуха. В случаях, описанных Rosenberg и Chutorian, потеря слуха отмечалась с детства. Полная глухота развилась в 5—6-летнем возрасте. Речь была развита слабо и оставалась рудиментарной. Аудиометрическое исследование мужчин сибсов в возрасте 29 и 32 лет обнаружило у них двустороннюю нейросенсорную глухоту от 60 до 90 дБ. Трехлетний мальчик реагировал только на громкие шумы. Другие аудиологическис тесты не описаны. У больных сибсов, изученных Iwashita с сотр., глубокая нейросепсорная глухота была обнаружена в возрасте около 15 лет.
Вестибулярная система. Результаты вестибулярных проб не представлены.
Лабораторные данные. Рентгенографическое, электроэнцефалографическое, электроретииографическое и электрокардиографическое исследования патологии не выявили. Электромиографическое исследование обнаружило у трех больных, описанных Rosenberg и Chutorian, редукцию нервной проводимости от легкой до умеренной степени. У сибсов, о которых сообщили Iwashita с сотр., были выявлены сколиоз и нормальный уровень проводимости в двигательных нервах.
Анализы крови и мочи, включая исследование уровня креатинин-фосфокиназы в сыворотке и спинномозговой жидкости, также были нормальными.
Патология. Биопсия икроножной мышцы у 2 старших больных, описанных Rosenberg и Chutorian, показала резко выраженную невральную атрофию. У младшего больного мышцы выглядели нормальными, но биопсия икроножной мышцы показала демиелинизацию с сохранностью аксонов. Исследования ультраструктуры мышц специфических изменений не выявили (Ohta, 1970).
Наследственность. В семье, описанной Rosenberg и Chutorian, у 2 братьев наблюдалось одинаковое течение болезни. Ранние симптомы заболевания были обнаружены и у их племянника. Наследование похоже на Х-сцепленное рецессивное. Однако в сибсовой паре брат—сестра, описанной Iwashita с сотр., более вероятен аутосомно-рецессивпый тип наследования. Возможно, эти два состояния представляют разные заболевания.
Диагноз. Синдром Шарко—Мари—Тута генетически гетерогенен и характеризуется мышечными атрофиями, начинающимися с мышц стоп и ног (Dyck, Lambert). Первыми вовлекаются перонеальные мышцы, позже поражаются передняя большеберцовая мышца, длинный разгибатель пальцев или икроножная мышца. Со временем процесс может охватить руки и плечи. Часто наблюдаются полая стопа и молоткоподобные I пальцы (Allen). Хотя атрофия зрительных нервов при синдроме Шарко—Мари—Тута встречается редко, в некоторых случаях она описана.
Атрофия перонеальных мышц может сочетаться с катарактой и глухотой (Campbell). Она может встречаться также при чувствительной корешковой нейропатии, доминантно наследуемом синдроме, заключающемся в выраженном нарушении периферической чувствительности, трофических язвах па ногах и нейросенсорной глухоте (Campbell, Hoffman). Dejerine и Thomas описали атрофии конечностей, осязательных нервных окончаний и нейросенсорпую глухоту у брата и сестры. Можно думать, что у этих больных был синдром, который здесь обсуждается.
Болезнь Лебера (наследственная атрофия зрительных нервов) генегически гетерогенна, но должна быть также обсуждена, ибо она выявляется в молодом возрасте и может сочетаться с атаксией, пирамидными нарушениями и симптомами поражения задних столбов спинного мозга. Иногда она может сочетаться и с дефектами слуха (Wilson). Однако в семье, описанной Veit, сочетание глухоты с леберовской атрофией зрительных нервов выглядит случайным.
Заболевание у сестер, о которых сообщали Jequier и Deonna, вероятно, представляет отдельный синдром. У обеих в возрасте от 8 до 10 лет обнаружена быстро прогрессирующая нейросенсорная глухота и вестибулярная дисфункция. Впоследствии, почти немедлнно вслед за этим, выявилась быстро прогрессирующая потеря зрения, развившаяся вследствие атрофии зрительных нервов. Несколько лет спустя были утрачены проприоцептивная чувствительность, вибрационная чувствительность и глубокие рефлексы. Кроме того, у обоих сибсов, была обнаружена болезнь позвоночника Шейерманна. Наследование, вероятно, было аутосомно-рецессивпым.
Лечение. В течение первых нескольких лет жизни могут быть полезны слуховые аппараты.
Прогноз. Прогноз для зрения и слуха неблагоприятный в связи с прогрессирующим течением болезни.
Выводы. Характеристика этого синдрома включает: 1) Х-сцепленное или аутосомно-рецессивное наследование; 2) прогрессирующую потерю зрения в связи с атрофией зрительных нервов, начинающуюся в возрасте около 20 лет; 3) прогрессирующую периферическую полинейропатию, начинающуюся в раннем детстве, и 4) прогрессирующую нсйросенсорную глухоту, приводящую к полной потере слуха к 6-летнему возрасту.
Наследственные полинейропатии
Наследственные полинейропатии
(информация для пациентов)
Что такое "Наследственные полинейропатии"?
Наследственные полинейропатии – это большая группа клинически разнообразных полинейропатий с возможным вовлечением различных органов и систем, в том числе и центральной нервной системы, которые развиваются в результате мутаций (поломок) в генах человека.
- "Наследственные заболевания развиваются с рождения или в раннем детстве"
Это не так - дебют наследственной болезни возможен и во взрослом возрасте, в том числе и после 50 лет. Более того, зачастую пациенты не могут точно назвать возраст начала заболевания, так как симптомы развиваются незаметно. - "Наследственная патология должна быть и у близких родственников. А если в роду ни у кого нет, значит и у меня тоже нет".
Это не так - наследственные заболевания передаются разными путями: по аутосомно-доминантному, аутосомно-рецессивному, Х-сцепленному типу, также возможен митохондриальный тип наследования и др. Часто в семье есть асимптомные носители мутантного гена без клинических признаков заболевания, а симптомы болезни могут быть только у одного из членов семьи. Поэтому отсутствие отягощенного семейного анамнеза не исключает наследственный генез заболевания.
- Наследственные моторно-сенсорные нейропатии (НМСН)
- Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС)
- Наследственные сенсорные и автономные нейропатии (НСАН)
- Болезнь Фабри
- Транстиретиновая семейная амилоидная нейропатия (ТТР-САП)
Как часто диагностируются наследственные полинейропатии среди населения?
Частота встречаемости всех форм НМСН варьирует от 10 до 40 случаев на 100 000 населения в различных популяциях. На НМСН 1 типа приходится до 70% всех случаев НМСН. Распространенность ННСПС 2-5 случая на 100 000 населения. Болезнь Фабри - 1 случай на 40 000 - 60 000 мужчин. По примерным подсчетам в США встречаемость ТТР-САП составляет 1 случай на 100 000 человек. Следует отметить, что настороженность относительно дебюта наследственной патологии во взрослом возрасте среди врачей разных специальностей достаточно низкая, поэтому некоторые цифры распространенности могут быть занижены.
- пациентов беспокоят болезненные спазмы мышц голеней (крампи), слабость и деформация стоп, изменение походки, затруднения при беге или подъеме по лестнице;
- постепенно слабость развивается в кистях, в результате чего появляются затруднения при застегивании пуговиц, открывании двери ключом и т.д. В целом руки вовлекаются не ранее чем через 10 лет после появления первых симптомов болезни;
- в меньшей степени беспокоит онемение кистей и стоп, часто эти изменения игнорируются пациентом.
- лучевой нерв на уровне плеча (спиральный канал), при этом развивается слабость разгибателей кисти, кисть "висит" как плеть, возникает онемение наружного края предплечья и кисти;
- малоберцовый нерв на уровне коленного сустава (фибулярный канал), при этом развивается слабость разгибателей стопы, стопа начинает "шлепать", возникает онемение наружного края голени и стопы;
- локтевой нерв на уровне локтевого сустава (кубитальный канал), при этом развивается слабость межкостных мышц кисти, мышцы - отводящих мизинец и других, возникает онемение 4 и 5 пальцев кисти;
- срединный нерв на уровне запястья (карпальный канал), при этом развивается слабость мышц возвышения большого пальца, возникает онемение 1-3 пальцев кисти, характерен болевой синдром;
- и т.д. с развитием соответствующей клинической картины поражения того или иного нерва.
Степень поражения нерва (защемления в канале) может быть различной - от незначительной, когда беспокоит только онемение и парестезии, до более выраженной, когда помимо чувствительных нарушений развивается и слабость мышц.
Развитию симптомов, как правило, предшествует незначительная травма, нахождение длительное время в неудобной статической поза (на четвереньках, на корточках, облокотившись на локоть и др.), ношение неудобной одежды, непривычная физическая нагрузка и прочее.
Мышечная слабость, как правило, сохраняется в течение нескольких дней или недель, а затем сила мышц постепенно восстанавливается (в течение нескольких недель или месяцев). Эпизоды "защемлений" нервов имеют тенденцию к повторению (всего у пациентов может быть от 1 до 10 таких эпизодов). Следует учитывать, что часто лечащие врачи не задумываются о том, что причиной рецидивирующих (повторяющихся) туннельных невропатий – является наследственная патология, что приводит к поздней диагностике и неоправданным медицинским вмешательствам (операциям по поводу туннельных невропатий).
Болезнь Фабри - редкое генетическое заболевание, обусловленное мутацией гена GLA, картированного на длинном плече хромосомы Хq 22.1 и контролирующего структуру фермента альфа-галактозидазы А. Нередко начало заболевания отмечается в подростковом возрасте. Тип наследования - сцепленное с Х-хромосомой.
- жгучая колющая боль в ладонях и стопах; острые приступы мучительной боли в кистях и стопах (кризы Фабри);
- нарушение потоотделения; непереносимость жары/холода;
- сыпь на коже (ангиокератомы);
- помутнение роговицы в виде завитка, которое не ослабляет зрение;
- шум в ушах, потеря слуха;
- желудочно-кишечные расстройства, диарея;
- кардиологические проявления (включая увеличенное сердце и нарушение ритма);
- нарушение функции почек, которое в конечном итоге приводит к терминальной стадии хронической почечной недостаточности;
- нарушение мозгового кровообращения (чаще ишемический инсульт, но может быть и внутримозговое кровоизлияние, или венозный тромбоз).
- анализ истории развития заболевания
- оценка неврологического статуса
- общий клинический и развернутый биохимический анализы крови;
- RW, анти-ВИЧ, НВsAg и анти-HCV;
- общий анализ мочи;
- анализ крови на уровни витаминов группы В, гомоцистеин;
- электрофорез белков сыворотки и мочи с иммунофиксацией; ;
- анализ цереброспинальной жидкости (ликвор) и другие.
- уровень фитановой кислоты в крови методом хроматомасс-спектрометрии (повышение при болезни Рефсума);
- определение активности лизосомного фермента альфа-галактозидазы А, в том числе с помощью флюориметрического метода ("сухое" пятно) - исключение болезни Фабри;
- биопсия икроножного нерва, малых слюнных желез и абдоминального жира – для выявления отложения амилоида, характерного для ТТР-САП).
- Однако окончательный диагноз той или иной наследственной полинейропатии может быть подтвержден только по результатам генетического анализа. Оптимальным является генетическое обследование по следующему алгоритму:
- при убедительной клинико-анамнестической и параклинической картине в пользу той или иной наследственной полинейропатии проводится прицельная генетическая диагностика одного гена, в том числе с использованием скрининг тестов методом "сухого пятна";
- если подозревается наследственный генез полинейропатии, тип её не определен целесообразно проведение более широкого генетического анализа - секвенирования нового поколения (NGS).
- при болезни Фабри - это фермент-заместительная терапия рекомбинантными препаратами альфа-галактозидазы А
- при ТТР-САП – стабилизация молекулы транстиретина (Тафамидис), трансплантация печени;
- при порфирийной полинейропатии - введение аргината гема;
- при болезни Рефсума – диетотерапия с ограничением поступления фитановой кислоты (исключение потребления зеленых овощей, говядины, рыбы (тунец, пикша, треска)), плазмаферез.
При некоторых наследственных нейропатиях, для которых характерно поражение органов и систем, важно мониторировать функцию сердечно-сосудистой системы и других органов, наблюдаться у профильных специалистов.
- рекомендуется вести здоровый образ жизни, правильно и сбалансировано питаться, полностью отказаться от приема алкоголя, воздерживаться от вегетарианства;
- исключить, ограничить прием лекарственных препаратов с нейротоксическим действием;
- проводится коррекция ортопедических нарушений: подбор стелек, стоподержателей, коленоупорников, использование дополнительной опоры, кинезиотейпирование;
- регулярно, в поддерживающем режиме проводятся реабилитационные мероприятия: миостимуляция, массаж, растяжка, ЛФК.
Прием "нейрометаболических" препаратов и витаминов группы В не целесообразно, в связи с отсутствием доказательной базы.
.
Какой прогноз данного заболевания?
Прогноз определяется типом наследственной полинейропатии и многими сопутствующими факторами. Так, при НМСН 1 и 2 типа при позднем дебюте крайне замедленное развитие мышечной слабости приводит к тому, что больные приспосабливаются к своему дефекту и, несмотря на слабость и похудание кистей и стоп, могут выполнять ручную работу, свободно передвигаться. Даже при многолетнем течении сохраняется способность к самостоятельной ходьбе. Наследственная нейропатия со склонностью к параличам от сдавления также имеет относительно благоприятный прогноз при соблюдении рекомендаций по ограничению длительных статических поз и снижению потенциальных провокаторов компрессий нервов в повседневной жизни. НМСН III, V типов вызывают тяжелый двигательный дефект и в большинстве случаев приводит к выраженной инвалидности. При ранней диагностике наследственных нейропатий, ассоциированных с метаболическим дефектом (ТТР-САП, болезнь Рефсума, порфирия, болезнь Фабри) и назначении необходимой терапии возможно пролонгировать наступление инвалидизации и улучшить качество жизни.
В ФГБНУ НЦН проводят консультации сотрудники Центра заболеваний периферической нервной системы, сотрудники 5 неврологического отделения, профилем которого является диагностика и лечение наследственных заболеваний нервной системы. Консультации пациентов проводятся амбулаторно в рамках ОМС и на коммерческой основе.
На базе ФГБНУ НЦН проводится весь перечень необходимых для уточнения диагноза обследований, разработаны подходы к комплексной восстановительной терапии при наследственных полинейропатиях.
Наследственная нейропатия зрительного нерва
Наследственные нейропатии зрительного нерва являются результатом генетических дефектов, которые вызывают снижение остроты зрения и иногда приводят к патологии сердца и нервной системы. Эффективного лечения не существует.
В группу наследственных нейропатий зрительного нерва входят доминантная атрофия зрительного нерва и наследственная нейропатия зрительного нерва Лебера; оба заболевания являются митохондриальными цитопатиями ( 1 Общие справочные материалы Наследственные нейропатии зрительного нерва являются результатом генетических дефектов, которые вызывают снижение остроты зрения и иногда приводят к патологии сердца и нервной системы. Эффективного. Прочитайте дополнительные сведения ). Эти заболевания обычно проявляются в детском или подростковом возрасте двусторонним симметричным выпадением центральных полей зрения. Повреждения зрительного нерва обычно носят постоянный характер, в некоторых случаях прогрессируют. К моменту диагностики атрофии зрительного нерва обычно уже развивается его стойкое поражение.
Доминантная атрофия зрительного нерва
Доминирующая атрофия зрительного нерва имеет аутосомно-доминантный тип наследования. Это заболевание считаетсясамой распространенной наследственной нейропатией (заболеваемость с частотой от 1:10 000 до 1:50 000 населения). Считается, что оптическая абиотрофия является причиной преждевременной дегенерации зрительного нерва, что приводит к прогрессирующей потере зрения. Манифестация наступает в первом десятилетии жизни.
Наследственная атрофия зрительного нерва Лебера
Наследственная атрофия зрительного нерва Лебера характеризуется дефектом митохондриальной ДНК Расстройства митохондриального окислительного фосфорилирования Нарушение окислительного фосфорилирования часто, но не всегда, приводит к молочному ацидозу, влияющему, в частности, на центральную нервную систему, сетчатку и мышцы. См. также Подходы к лечению. Прочитайте дополнительные сведения , что нарушает клеточное дыхание. Несмотря на то что страдают ДНК митохондрий всех клеток тела, первым проявлением болезни является именно потеря зрения. Чаще всего это заболевание наблюдается у мужчин (80–90% случаев). Болезнь наследуется по женской линии, nо есть все потомки женщины с патологическими генами унаследуют патологию, но только женщины могут передать ее дальше, потому что зигота получает митохондрии только от матери.
Общие справочные материалы
1. Kisilevsky E, Freund P, Margolin E: Mitochondrial disorders and the eye. Surv Ophthalmol 65(3):294-311, 2020. doi: 10.1016/j.survophthal.2019.11.001
Симптомы и признаки наследственных нейропатий зрительного нерва
Доминантная атрофия зрительного нерва
Наследственная атрофия зрительного нерва Лебера
У некоторых пациентов с наследственной атрофией зрительного нерва Лебера также наблюдаются нарушения проводимости сердца. У других пациентов наблюдаются малые неврологические отклонения, такие как постуральный тремор, потеря лодыжечного рефлекса, дистония, спастичность или заболевания, схожие с рассеянным склерозом.
Наследственная Оптическая Нейропатия Лебера

Наследственная Оптическая Нейропатия Лебера (NOHL или LHON), также известная как Атрофия Зрительного Нерва Лебера (LOA), названа в честь доктора Теодора Лебера, который описал её в 1871 году, как характерную картину внезапной потери зрения у молодых людей, в с семейном анамнезе которых наблюдалась потеря зрения. Это самая распространенная наследственная оптическая невропатия, её причиной является митохондриальная мутация и её распространенность очень низкая, их в большинстве географических районов она неизвестна. На северо-востоке Англии и Финляндии заболевание затрагивает 1 человека из каждых 31 000 или 50 000 соответственно.
Это обычно наблюдается у молодых мужчин в возрасте от 18 до 35 лет, хотя это может также повлиять на детей младшего возраста и взрослых старше 35 лет. У женщин оно встречается гораздо реже.
Как правило, оно вызывает серьезную потерю зрения в обоих глазах. В большинстве случаев, процесс начинает в одном глазу и через несколько недель или месяцев переходит на второй глаз.
Почему оно производится? Что такое наследственность митохондрий?
Хотя подавляющее большинство нашего генетического материала (ДНК) расположено в ядре клеток, его небольшая часть находится в митохондриях (митохондриальная ДНК или мтДНК).
Гены ядра наследуются от обоих биологических родителей. Однако, ДНК митохондрии наследуются только от матери. Это означает, что мужчина с мутацией в митохондриальной ДНК не передаст его ни одному из своих биологических потомков, тогда как женщина с мутацией в митохондриальной ДНК передаст её всем своим биологическим потомкам.
90-95% случаев LHON связаны с одной из трех специфических мутаций мтДНК. Однако значительная часть людей с этими мутациями никогда не развивают симптомы заболевания. В частности, только 10% женщин и 50% мужчин, несущих любую из этих мутаций, имеют оптическую нейропатию. Это подразумевает, что другие факторы, генетические (митохондриальные или ядерные) и / или факторы окружающей среды должны действовать так, чтобы произошла потеря зрения. Так, например, воздействие табака или большого количества алкоголя у людей, несущих эти мутации, увеличивает риск развития заболевания.
Симптомы Оптической Нейропатии Лебера
Как правило, носители LHON не имеют до тех пор, пока они не теряют значительно быстро зрение в одном глазу и через несколько недель или месяцев также в другом глазу. Зрение продолжает ухудшаться в течение нескольких недель, пока оно не станет значительным, и в большинстве случаев центральное видение (необходимое для чтения, вождения и распознавания лиц) серьезно страдает в обоих глазах, хотя некоторые люди испытывают определенное восстановление зрения через некоторое время. Эта постоянная потеря зрения является следствием смерти клеток зрительного нерва, ответственных за передачу изображений из глаз в головной мозг.
Хотя потеря зрения является единственным симптомом у большинства пациентов с LHON, в некоторых случаях из них были описаны нарушения сердечного ритма, а также неврологические изменения (такие как постуральный тремор или другие нарушения движения).
Как оно диагностируется? Существует лечение этого заболевания?
Это заболевание сложно диагностировать по нескольким причинам. Во-первых, это наследственное заболевание, но оно не проявляется у всех людей, имеющих мутации. По этой причине он может появиться без учета семейного анамнеза болезни.
Чтобы диагностировать его, обычно необходимо провести тщательную нейрофтальмологическую оценку и анализ крови для исследования мтДНК.
В анализе глазного дна сначала наблюдается микроангиопатия и отек слоя перипапиллярного нервного волокна, который прогрессирует до оптической атрофии.
В настоящее время прогноз обычно представляет собой постоянную серьезную потерю зрения. Изучается несколько способов лечения этого заболевания, и в ICR проводится исследование болезни, которая находится на этапе отбора.
Читайте также: