Варианты патологического гемоглобина. Гемоглобинопатии
Добавил пользователь Дмитрий К. Обновлено: 23.01.2026
Механизмы развития гемоглобинопатий - патогенез, классификация
Отдельные структурные изменения аномальных гемоглобинов по разному влияют на физико-химические свойства, устойчивость и функцию гемоглобина. В значительной мере это зависит от природы структурного сдвига и уровня, на котором происходит аномалия в трехмерном строении молекулы гемоглобина.
Многие из аномальных гемоглобинов не проявляются клиническими признаками заболевания, поскольку биохимическое изменение в той или иной «несущественной» области молекулы лишь в незначительной мере или совсем не влияет на стойкость и функцию гемоглобина.
Однако замена какой-либо аминокислоты в определенной «критической » области одной из цепей может отрицательно повлиять на целостность и функцию молекулы в целом, при этом клиническая реакция в значительной мере будет отражать структурную аномалию.
В принципе структурные изменения на поверхности молекулы не влияют на свойства гемоглобина и не вызывают каких-либо клинических последствий. Однако к этой категории относятся и некоторые гемоглобины, структурное изменение которых вызывает глубокие сдвиги физических свойств молекулы, которые, в клиническом плане, проявляются тяжелыми гемолитическими анемиями. Одним из таких примеров служит гемоглобин S.
В нем изменение пространственной конфигурации цепи bs в безкислородной форме обусловливает образование межмолекулярынх связей с цепями ос соседних молекул, а это составляет основу аггрегатов гемоглобина и сиклизации гематий. Другим примером той же категории служит гемоглобин С; заниженная растворимость этого гемоглобина составляет причину изменения формы эритроцитов.
В сохранении устойчивости гемоглобина и нормальной функции переноса О2 особое значение представляют межцепные связи. Структурные аномалии у контакта а1в1г обусловливают утрату молекулярной целостности и развитие гемолитической анемии, характерной отдельным нестойким гемоглобинам (напр. гемоглобин Тасота и гемоглобин Philly).
Обычно внутримолекулярные структурные сдвиги, в частности в отдельных «критических» областях, приводят к неустойчивости молекулы, оседанию в эритроците гемоглобинных подъединиц и образованию телец Гейнца. Связи цепей а1в1 способствуют сохранению явления взаимодействия «гем-гем» и функции переноса О2. Структурные сдвиги на этом уровне изменяют сродство гемоглобина к О2 и его дыхательную функцию (напр. гемоглобин Kempsey, гемоглобин Chesapeake).
Замена аминокислот у места связывания гема с глобином приводит к образованию стойкой связи между «аномальной » аминокислотой и железом гема, при этом Fe2+ преобразуется в Fe3+ и развитию метгемоглобинемии.
В заключении можно утверждать следующее:
а) с клинической точки зрения различные аномальные, известные до настоящего времени, гемоглобины могут обусловить ряд важных синдромов, в том числе, гемолитическую анемию, врожденный цианоз, семейную полицитемию, синдромы, похожие на симптомокомплекс талассемии;
б) клинические аспекты находятся в тесной взаимосвязи с природой и уровнем структурной аномалии в комплексном пространственном строении молекулы гемоглобина.
Ниже приведена схема физиопатогенетической классификации заболеваний гемоглобина, за счет его аномалий.
| Клиническая картина | Гемоглобин | Структурный недостаток | Последствия структурной аномалии |
| I. Гемоглобинопатия в сочетании с гемолитической анемией | Гемоглобин S, гемоглобин С, Гемоглобин Е, гемоглобин D Гемоглобин Zurich Гемоглобин Koln Гемоглобин Hammer-schmith Гемоглобин Gun Hill и т.д. | Снаружи молекулы: — Карман гема — Контакт a1b1 — Пространственная конфигурация цепей глобина | Искажение эритроцита Неустойчивость гемоглобина Образование телец Гейнца |
| II. Гемоглобинопатии в сочетании с наследственной метгемоглобинемией | Гемоглобин М | — Прикрепление гема к глобину | Метгемоглобинизация |
| III. Гемоглобинопатия в сочетании с семейной полицитемией | Гемоглобин Capetown Гемоглобин Yashima Гемоглобин Chesapeake и т.д. | Контакт а1b1 Место связывания 2—3 дифосфоглицерата | Рост сродства к О2 |
| IV. Гемоглобинопатия в сочетании с гемолитической анемией и синюхой | Гемоглобин Kansas | Карман гема | Понижение сродства к О2 |
| V. Гемоглобинопатия в сочетании с клиническим аспектом талассемического синдрома | Гемоглобин Lepore Гемоглобин Constant Spring Гемоглобин Icaria Гемоглобин Коуа Dora | Слияние цепей Гемоглобины удлиннеными цепями | Сокращение коэффициента синтеза гемоглобина |
Диагноз гемоглобинопатии подсказывают данные анамнеза (течение болезни наблюдается уже с детства, наличие одинаковых признаков и у других членов семьи), физические признаки (желтуха или поджелтушное состояние, спленомегалия, цианоз и пр.) и лабораторные исследования (выявляющие анемию, признаки гемолиза, морфологические изменения эритроцитов и пр.).
Клиническое и гематологическое исследование необходимо пополнить специальными обследованиями, способными выявить аномалию гемоглобина. Некоторые из них составлены из простых методов, применение которых доступно любой обычно оснащенной клинической лаборатории, в том числе, реакция сиклизации, определение шелочноустойчивого гемоглобина, окраска для выявления телец Гейнца, тест на тепловую устойчивость и пр.
Все же основным методом в постановке диагноза гемоглобинопатии представляется электрофорез гемоглобина, хотя следует незабывать его пределы (аномальный гемоглобин с похожей на гемоглобин А миграцией, случаи талассемии b и а с электрофорезом в норме и пр.). Способ «fingerprint» и анализ аминокислот, способствующих уточнению структурной аномалии, применяются лишь в специализированных лабораториях. Семейная разведка облегчает диагностирование и уточняет способ наследственной передачи.
Толкование результатов всех отмеченных методов требует опыт специалиста гематолога и сопоставление с полным клиническим и гематологическим обследованием больного.
Течение гемоглобинопатий. В принципе при гетерозиготных формах прогноз хороший и жизнь] больных протекает почти нормально, за исключением отдельных случаев талассемии, при которых может развиться тяжелая гемолитическая анемия. Гетерозиготные формы дрепаноцитоза и талассемии обычно характеризуются тяжелым прогнозом и лишь в небольшом числе случаев больные достигают зрелого возраста.
У гомозиготов в отношении гемоглобина С, Е и D заболевание протекает обычно по виду гемолитической анемии средней тяжести.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Метгемоглобинемия
Метгемоглобинемия – состояние, характеризующееся повышенным содержанием метгемоглобина (окисленного гемоглобина) в крови и тканевой гипоксией. Развитие метгемоглобинемии сопровождается акроцианозом, слабостью, головными болями, головокружением, сердцебиением, одышкой при нагрузке. Характерным признаком метгемоглобинемии служит коричнево-шоколадный цвет крови. Для подтверждения диагноза проводится оценка симптоматики, лабораторные исследования и тесты. При тяжелой степени метгемоглобинемии показана кислородотерапия, введение аскорбиновой кислоты, раствора метиленового синего, в ряде случаев - обменная гемотрансфузия.
МКБ-10
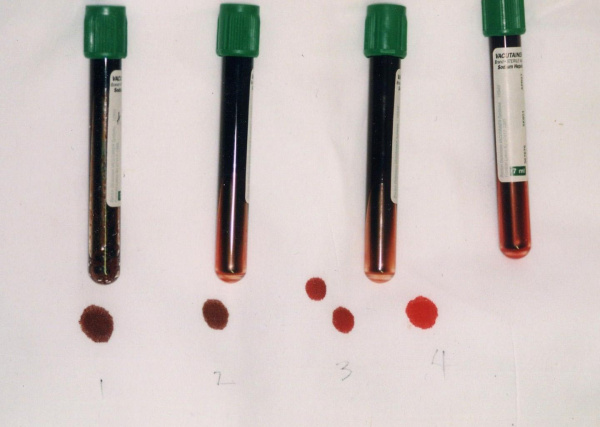
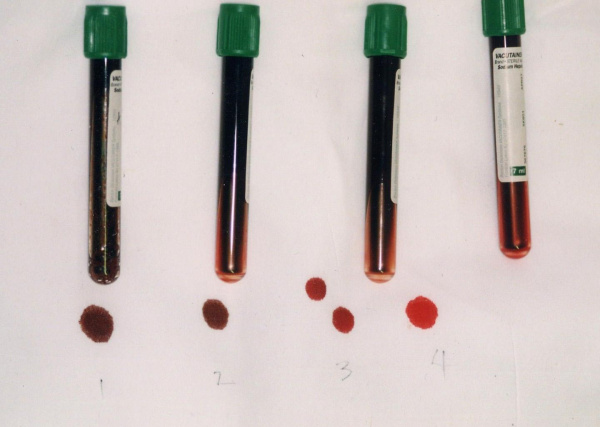
Общие сведения
Метгемоглобинемия – увеличение уровня гемоглобина, содержащего окисленное железо (метгемоглобин – MtHb), в эритроцитах крови. Метгемоглобин относится к так называемым дисгемоглобинам - дериватам гемоглобина, не способным транспортировать кислород. В обычных условиях в крови присутствует небольшое количество метгемоглобина - не более 1% от общего содержания Hb. При метгемоглобинемии эндогенные механизмы оказываются не способными регулировать концентрацию дисгемоглобина, в результате чего страдает кислородно-транспортная функция эритроцитов. В гематологии метгемоглобинемии подразделяются на наследственные и приобретенные. Первые из них распространены среди коренного населения Аляски, Гренландии, Якутии; частота развития приобретенных метгемоглобинемий неизвестна.
Причины метгемоглобинемии
В процессе метаболических превращений в крови здоровых людей в очень небольших количествах образуются дисгемоглобины: карбоксигемоглобин, сульфгемоглобин, метгемоглобин (0,1-1 %). Вместе с тем, в эритроцитах содержится ряд факторов, которые поддерживают долю фракции метгемоглобина на уровне, не превышающем 1,0-1,5% от общего Hb. В частности, в реакции восстановления метгемоглобина в гемоглобин участвует фермент метгемоглобин-редуктаза. В отличие от оксигемоглобина (НbО2), содержащего восстановленное железо (Fe++), в метгемоглобине содержится окисленное железо (Fe+++), не способное переносить кислород. Поэтому при метгемоглобинемии, прежде всего, страдает кислородно-транспортная функция крови, следствием которой служит тканевая гипоксия.
Наследственные формы метгемоглобинемии представлены либо ферментопатиями (врожденной низкой активностью или отсутствием фермента метгемоглобин-редуктазы), либо М-гемоглобинопатиями (синтезом аномальных белков, содержащих окисленное железо).
В структуре приобретенных (вторичных) метгемоглобинемий выделяют токсические экзогенные и токсические эндогенные формы. Метгемоглобинемии экзогенного происхождения могут быть связаны с передозировкой лекарственных средств (сульфаниламидов, нитритов, викасола, противомалярийных препаратов, лидокаина, новокаина и др.) или отравлением химическими агентами (анилиновыми красителями, нитратом серебра, тринитротолуолом, хлорбензолом, питьевой водой и пищевыми продуктами с высоким содержанием нитратов и др.).
Повышенный уровень MtHb в крови наблюдается у недоношенных и доношенных новорожденных, что связано с низкой активностью фермента метгемоглобин-редуктазы и окислительным стрессом в родах. Однако даже при тяжелой гипоксии и желтухе новорожденных подъем MtHb не столь выражен и клинически значим, чтобы послужить причиной метгемоглобинемии. Однако при диарее, бактериальных и вирусных энтероколитах, в условиях метаболического ацидоза у детей первого года жизни может легко развиться приобретенная эндогенная метгемоглобинемия.
О смешанной форме патологии говорят в том случае, если метгемоглобинемия развивается под воздействием экзогенных факторов у здоровых лиц, являющихся гетерозиготными носителями генов наследственной формы заболевания.
Симптомы метгемоглобинемии
Признаки наследственной метгемоглобинемии становятся заметны в период новорожденности. На коже и видимых слизистых ребенка (в области губ, носогубного треугольника, мочек ушей, ногтевого ложа) заметен цианоз. Кроме наследственной метгемоглобинемии, у детей часто выявляются другие врожденные аномалии - изменения конфигурации черепа, недоразвитие верхних конечностей, атрезия влагалища, талассемия и пр. Нередко дети отстают в психомоторном развитии.
В зависимости от уровня фракции MtHb, выраженность проявлений врожденной и приобретенной метгемоглобинемии может значительно варьировать.
При концентрации MtHb в крови:
Для любых форм метгемоглобинемии характерна грифельно-серая окраска кожных покровов, однако отсутствуют характерные для сердечно-легочных заболеваний изменения ногтевых фаланг по типу «барабанных палочек». Акроцианоз усиливается при охлаждении, употреблении в пищу нитратосодержащих продуктов, при токсикозах беременности у женщин, а также приеме метгемоглобинобразующих медикаментов.
Диагностика метгемоглобинемии
Важным диагностическим признаком метгемоглобинемии служит темно-коричный цвет крови, которая, будучи помещенной в пробирку или на фильтровальную бумагу, не изменяет свой цвет на красный. При положительной пробе проводится спектроскопия, определение концентрации MtHb, активности НАД-зависимой метгемоглобинредуктазы, электрофорез гемоглобина.
В общем анализе крови может присутствовать компенсаторный эритроцитоз, увеличение Hb, ретикулоцитоз, уменьшение СОЭ. При исследовании биохимических показателей крови определяется незначительная билирубинемия, обусловленная увеличением непрямой фракции пигмента. Для хронической метгемоглобинемии типично появление в эритроцитах телец Гейнца-Эрлиха.
У больных с энзимопенической или токсической метгемоглобинемией показательна терапевтическая проба с внутривенным введением метиленового синего – после инъекции цианоз быстро исчезает, а кожа и видимые слизистые приобретают розовую окраску.
При анализе причин метгемоглобинемии важно выяснить, имел ли больной контакт с токсическими веществами, принимал ли метгемоглобинобразующие лекарственные препараты. При подозрении на врожденную метгемоглобинемию изучается родословная, проводится консультация генетика, определяется тип наследования патологии крови. Наследственные метгемоглобинемии требуют дифференциации с врожденными пороками сердца синего типа, аномалиями развития легких и другими состояниями, сопровождающимися гипоксией.
Лечение и профилактика метгемоглобинемии
Больные с отсутствием клинических проявлений не нуждаются в специальной терапии. При значительной концентрации MtHb в крови и развернутой симптоматике метгемоглобинемии назначается медикаментозная терапия, способствующая превращению метгемоглобина в гемоглобин. Такими восстанавливающими свойствами обладают аскорбиновая кислота и метиленовый синий. Аскорбиновая кислота назначается внутрь сначала в больших, а по мере нормализации состояния - в поддерживающих дозах. Раствор метиленовой сини вводится внутривенно. При выраженном цианозе проводится кислородная терапия. Тяжелая форма метгемоглобинемии является показанием к обменному переливанию крови.
Течение наследственной и лекарственной метгемоглобинемии, как правило, доброкачественное. Неблагоприятный исход возможен при тяжелых формах токсической метгемоглобинемии с высоким содержанием MtHb в эритроцитах. Пациентам с подобной патологией следует избегать контакта с метгемоглобинобразующими веществами, переохлаждений и других провоцирующих факторов. Профилактика врожденной метгемоглобинемии заключается в проведении медико-генетической консультации для выявления гетерозиготных носителей среди будущих родителей.
Варианты патологического гемоглобина. Гемоглобинопатии
Наследственные нарушения гемоглобина можно подразделить на три большие группы, в зависимости от того, изменяет мутация белок глобина, его синтез или переключение глобинов:
• структурные варианты, изменяющие полипептид глобина, не влияя на показатели синтеза;
• талассемии, когда снижается синтез (или, редко, имеется крайняя нестабильность) одной или более цепей глобина, что приводит к дисбалансу относительных количеств а- и b-цепей;
• наследственное персистирование фетального гемоглобина, группа клинически благоприятных состоянии, представляющих интерес, поскольку они нарушают внутриутробное переключение синтеза от у-глобина к b-глобину.
Различные гемоглобины произошли вследствие точковых мутаций в одном из структурных генов глобина, но некоторые сформировались благодаря другим, более сложным молекулярным механизмам. Описано более 400 аномальных гемоглобинов, приблизительно половина их клинически значима.
Структурные варианты гемоглобина можно разделить на три класса, в зависимости от клинического фенотипа.
• Варианты, вызывающие гемолитическую анемию. Подавляющее большинство мутантных гемоглобинов, вызывающих гемолитическую анемию, приводят к неустойчивости тетрамера гемоглобина. Тем не менее два самых известных варианта, связанных с гемолизом, серповидно-клеточный гемоглобин (HbS) и НbС, не связаны с неустойчивостью, а заставляют мутантные белки глобина принимать необычную ригидную форму.
• Варианты с измененным транспортом кислорода вследствие повышенного или сниженного сродства к кислороду или образования метгемоглобина, формы глобина, неспособного к обратимому обогащению кислородом.
• Варианты, вызванные мутациями в кодирующем регионе, приводящими к талассемиям, за счет уменьшения количества полипептида глобина. Большинство этих мутаций нарушает скорость синтеза мРНК или белка. Некоторые редкие варианты вызывают общую нестабильность мономера гемоглобина, большую, чем при вариантах, ведущих к гемолитической анемии.
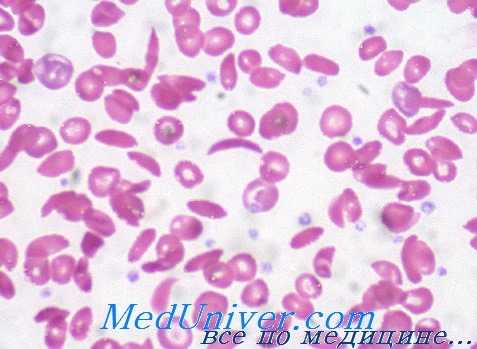
Структурные изменения выбраны для описания в данной статье, либо потому что они часто встречаются и типичны для одной из трех групп, либо иллюстрируют драматические биохимические и клинические последствия мутаций.
Варианты патологического гемоглобина:
1. HbS. Замена единственного нуклеотида. b-цепь: Glu6Val. Полимеризация ненасыщенного кислородом HbS -> серповидноклеточность —> закупорка сосудов и гемолиз. Аутосомно-рецессивное наследование.
2. HbC. Замена единственного нуклеотида. b-цепь: Glu6Lys. Насыщенный кислородом НbС стремится кристаллизоваться -> меньшая деформация клеток —> легкий гемолиз. Аутосомно-рецессивное наследование. Болезнь у компаундных гетерозигот HbS/HbC напоминает легкую серповидноклеточную анемию
3. Гемоглобин Hammersmith. Замена единственного нуклеотида. b-цепь: Phe42Ser. Неустойчивый гемоглобин -> преципитация гемоглобина —> гемолиз; также снижение сродства к кислороду. Аутосомно-доминантное наследование.
4. Hyde Park HbM. Замена единственного нуклеотида. b-цепь: His92Tyr. Замена делает окисленное железо гема устойчивым к редуктазе метгемоглобина —>HbM, не способный переносить кислород ->цианоз (бессимптомный). Аутосомно-доминантное наследование.
5. Гемоглобин Kempsey. Замена единственного нуклеотида. b-цепь: Asp99Asn. Замена удерживает гемоглобин в форме высокого сродства к кислороду —> снижение кислорода в тканях —> полицитемия. Аутосомно-доминантное наследование.
6. HbЕ. Замена единственного нуклеотида. b-цепь: Glu26Lys. Мутация —> аномальная структура гемоглобина и уменьшение его синтеза (аномальный сплайсинг РНК) —> мягкая талассемия. Аутосомно-рецессивное наследование.
Гемоглобинопатии
Гемоглобинопатии – это группа тяжелых наследственных заболеваний крови, обусловленных нарушением структуры гемоглобина или снижением синтеза одной и более глобиновых цепей. Клиническая картина крайне разнообразна. Общими симптомами являются гемолитическая анемия, увеличение селезенки, поражение костей. Диагностика осуществляется с помощью микроскопии мазка периферической крови, электрофореза гемоглобина, генетических исследований. Для лечения применяют переливание компонентов крови, препараты гидроксимочевины, инфузионную терапию. У тяжелых пациентов выполняется спленэктомия, аллотрасплантация стволовых клеток.
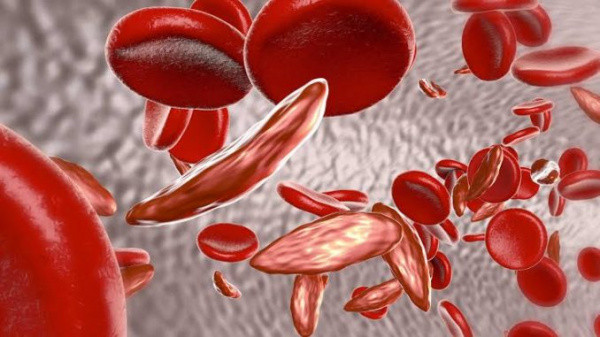
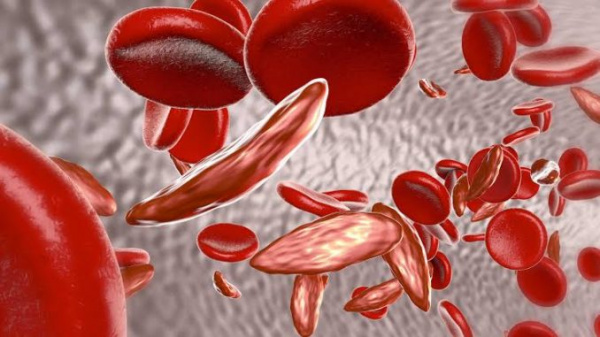
Гемоглобинопатии – ряд врожденных гемолитических анемий, характеризующихся изменением аминокислотной последовательности гемоглобина или подавлением образования цепей глобина. Данные патологии нередко заканчиваются летально уже в раннем детском возрасте. Известно около 50 видов гемоглобинопатий. Самыми частыми и опасными для жизни считаются серповидно-клеточная анемия (СКА), талассемии. Гемоглобинопатии распространены на территории Центральной Африки, Южной Азии и наблюдаются преимущественно у лиц негроидной расы. Талассемии также встречаются в странах Средиземноморья. Ежегодно рождается около 350000 детей с дефектами гемоглобина.
Причины гемоглобинопатий
Гемоглобинопатии относятся к аутосомно-рецессивным генетическим заболеваниям. Качественные гемоглобинопатии развиваются вследствие мутаций генов, ответственных за синтез определенных аминокислот в бета-цепи глобина. В результате происходит замена одной аминокислоты на другую (глутаминовой кислоты на валин, лизин и пр.). Это приводит к образованию аномального гемоглобина, гораздо менее растворимого, чем нормальный гемоглобин А, придающего красным кровяным тельцам иную форму (мишеневидную, серповидную), что нарушает их функции и уменьшает продолжительность жизни.
Количественные гемоглобинопатии обусловлены мутацией генов, которые кодируют целую цепь глобина (чаще альфа и бета). При этом сдвигается баланс между глобиновыми цепями – при недостаточном синтезе альфа-цепей возникает избыток бета-цепей, и наоборот. Уменьшаются размеры эритроцитов, в них снижается содержание гемоглобина, а мембрана становится более подверженной различным повреждениям.
Существуют факторы, провоцирующие тяжелые приступы (кризы). К ним относятся обезвоживание, переохлаждения, инфекции, сопровождающиеся высокой лихорадкой. У женщин обострения нередко развиваются на фоне беременности. Но главный патологический стимул качественных гемоглобинопатий – уменьшение концентрации в крови кислорода (гипоксия). Это может произойти, например, при подъеме на большую высоту (восхождение на гору, полет на самолете), где снижено парциальное давление кислорода воздуха, или при тяжелых болезнях дыхательной системы (пневмонии).
Патогенез
Гемоглобинопатии имеют сходные патогенетические механизмы. Измененная структура гемоглобина предрасполагает к интенсивному гемолизу. Длительно текущая анемия способствует компенсаторной гиперплазии костного мозга. Возникает деформация костей черепа, искривление позвоночника. Развиваются экстрамедуллярные очаги кроветворения, приводящие к увеличению размеров печени и селезенки (гепатоспленомегалии). Вследствие спленомегалии наступает гиперспленизм – усиленная деструкция красных клеток крови синусоидами селезенки.
Из-за регулярного гемолиза эритроцитов печень секретирует в желчь большое количество билирубина, что создает условия для образования камней желчного пузыря. У больных гемоглобинопатиями часто возникает перегрузка железом, как за счет постоянных переливаний крови, так и из-за повышения абсорбции железа желудочно-кишечным трактом. Большое количество железа в тканях усиливает процессы перекисного окисления липидов, что повреждает различные органы.
При качественных гемоглобинопатиях под влиянием сниженного содержания кислорода в крови молекулы нерастворимого аномального гемоглобина растягивают мембрану эритроцитов, что приводит к изменению их формы. Деформированные эритроциты хуже переносят кислород, а также способны приклеиваться к сосудистому эндотелию, тем самым закупоривая мелкие сосуды, вызывая тромбозы, окклюзии и инфаркты.
Классификация
Гемоглобинопатии подразделяются на качественные, обусловленные нарушением структуры (последовательности аминокислот) гемоглобина, и количественные, характеризующиеся снижением образования глобиновых цепей. Качественные гемоглобинопатии представлены следующими формами:
- Серповидно-клеточная анемия (Гемоглобинопатия S). Наиболее частый вид. Подразделяется на гомозиготную форму (собственно СКА) с яркой клинической симптоматикой и гетерозиготное носительство (серповидно-клеточную аномалию), имеющее бессимптомное или легкое течение.
- Гемоглобинопатия С. Клиника схожа с СКА, но менее выражена. Отличается большей степенью спленомегалии, чем при СКА.
- ГемоглобинопатияCS (Африканский ревматизм). По течению напоминает СКА. Преобладают приступообразные костно-суставные боли.
- Гемоглобинопатии Е иD. Протекают с небольшой гемолитической анемией.
- Наследственная метгемоглобинемия. При этой разновидности под влиянием различных факторов образуется окисленная форма гемоглобина – метгемоглобин, который более стойко связывается с кислородом и не отдает его тканям.
- Анемия, вызванная носительством нестабильных гемоглобинов. Это доброкачественная патология, при которой возникает незначительная гемолитическая анемия после приема сульфаниламидных препаратов.
К количественным аномалиям гемоглобина относят:
- Бета-талассемии. Наиболее распространенный вариант. Подразделяется на малую талассемию (гетерозиготное носительство с бессимптомным течением или легкой гемолитической анемией) и большую талассемию (анемию Кули) с развернутой тяжелой клинической картиной.
- Альфа-талассемии. Течение может быть различным в зависимости от количества мутантных генов. В основном сходны с гетерозиготной бета-талассемией.
- Синдром водянки плода c гемоглобином Барт (Hb Bart’s). Наиболее тяжелый вид альфа-талассемии. Ребенок погибает внутриутробно.
- ГемоглобинопатиюH. Благоприятная малосимптомная форма альфа-талассемии.
- Бета-дельта-талассемии. Практически неотличима от бета-талассемии.
- ГемоглобинопатиюLepore. Эта форма развивается вследствие слияния бета-цепей глобина и схожа с бета-талассемией.
Симптомы гемоглобинопатий
Клиническая картина гемоглобинопатий разнообразна. Гетерозиготные больные имеют либо бессимптомное, либо легкое течение. Гомозиготные формы начинают себя проявлять уже с раннего детства (6 месяцев - 1 год). Общая симптоматика включает признаки гемолитической анемии (бледность, желтушность кожи и слизистых, увеличение селезенки), патологию развития скелета – башенный череп (четырехугольный), уплощенная переносица, искривленный позвоночник.
Из-за повышенной секреции билирубина в желчь могут беспокоить симптомы желчнокаменной болезни уже в детском возрасте – тяжесть или ноющие боли в правом подреберье, приступы желчной колики, обесцвечивание кала. На коже голеней часто возникают длительно незаживающие язвы. Для болезней с дефектом в структуре гемоглобина характерно кризовое течение. Самые тяжелые приступы, нередко заканчивающиеся летально, встречаются при серповидно-клеточной анемии.
Вазоокклюзивный криз
Наиболее типичный. Происходит закупорка мелких сосудов различных органов. Дети испытывают боли в длинных трубчатых костях, у них отекают кисти, стопы, что затрудняет движения лучезапястных, голеностопных суставов (hand-foot синдром). Микротромбозы сосудов кишечника вызывают абдоминальные боли. У молодых мужчин нередко развивается приапизм, впоследствии приводящий к эректильной дисфункции. Кризу сопутствует лихорадка, тахикардия, потливость.
Гемолитический криз
При гемолитическом кризе происходит массивное разрушение красных телец с резким снижением содержания гемоглобина, эритроцитов в крови. Усиливается имеющаяся желтушность, бледность кожных покровов, слизистых, возникают лихорадка, поясничные и абдоминальные боли, присоединяются симптомы снижения артериального давления (головокружение, обмороки). Моча приобретает темный цвет за счет большого количества гемоглобина.
Секвестрационный криз
Во время секвестрационного криза также стремительно падает уровень гемоглобина. Но это происходит не из-за гемолиза, а вследствие венозного тромбоза и скопления большого количества крови в расширенных синусоидах печени и селезенке, что и приводит к обеднению общего кровотока. Кроме симптомов тяжелой анемии по причине выраженной гепатоспленомегалии появляются сильные и распирающие боли в левом и правом подреберье.
Апластический криз
Достаточно редкая форма криза. Она развивается при инфицировании парвовирусом В19, который способен угнетать костномозговое кроветворение. Это сопровождается стремительным (но обратимым) снижением концентрации не только эритроцитов, но и всех других клеток периферической крови (лейкоцитов, тромбоцитов). Поэтому к признакам анемии присоединяется геморрагический синдром (кровотечения из носа, десен), различные инфекции (в основном ОРВИ).
Осложнения
Общими осложнениями гемоглобинопатий считаются ЖКБ, патологические переломы длинных трубчатых костей. Гетерозиготные формы редко сопровождаются неблагоприятными событиями, так как имеют легкое течение. При количественных гемоглобинопатиях из-за отложения избыточного количества железа во внутренних органах развивается сердечная недостаточность, цирроз печени, сахарный диабет 2 типа.
Качественные патологии гемоглобина характеризуются широким спектром неблагоприятных последствий. Наиболее опасными считаются эмболия легочных сосудов, инфаркт миокарда, ОНМК, которые примерно у 10% пациентов приводят к смерти. Закупорка микрососудов, питающих кости, ведет к асептическому некрозу головки бедренной кости (АНГБК). Вследствие постоянных инфарктов селезенки возникает функциональный асплезнизм, из-за чего часто развиваются бактериальные инфекции (бронхиты, пневмонии) с тяжёлым течением, нередко с летальным исходом.
Диагностика
Курацией больных с гемоглобинопатиями занимаются врачи-гематологи, генетики. Во время общего осмотра обращается внимание на цвет кожных покровов (бледность, желтуха), конституциональные нарушения (задержку нервно-психического, физического развития ребенка, аномалии строения скелета). Дополнительное обследование включает:
Гемоглобинопатии дифференцируют с другими врожденными гемолитическими анемиями (мембранопатиями, ферментопатиями, микросфероцитарной анемией Минковского-Шоффара). Постоянные тромбозы нужно дифференцировать от различных тромбофилий. Перегрузку железом следует отличать от наследственного гемохроматоза. Анемия, оссалгии требуют исключения злокачественных миелопролиферативных заболеваний.
Лечение гемоглобинопатий
Людям, страдающим гемоглобинопатиями, требуется проведение сложной многокомпонентной терапии, поэтому все пациенты с гомозиготными формами подлежат обязательной госпитализации в гематологический стационар. Больным с гетерозиготными формами лечение не показано. Основные принципы ведения качественных и количественных патологий гемоглобина несколько отличаются друг от друга.
Консервативная терапия
Подбор консервативной терапии производится с учетом вида гемоглобинопатии, течения заболевания, наличия тех или иных осложнений. Оценивается как клиническая симптоматика, так и лабораторные данные (главным образом, показатели красной крови). Выделяют следующие направления лечения:
- Купирование кризов. При вазоокклюзивных кризах используются обезболивающие препараты (нестероидные противовоспалительные средства), а при выраженном болевом синдроме - наркотические анальгетики. Также для подавления преципитации деформированных эритроцитов назначаются ингаляции кислородом, пероральная или внутривенная регидратация.
- Предупреждение кризов. Для постоянного приема больным качественными гемоглобинопатиями назначается гидроксимочевина. Этот препарат стимулирует образование фетального гемоглобина, подавляющего экспрессию гена, ответственного за синтез аномальных нерастворимых гемоглобинов, что уменьшает склонность эритроцитов к деформации, снижает частоту кризов.
- Терапия осложнений. Инфекционные осложнения до получения результатов бактериологических исследований лечат антибактериальными препаратами, активными против пневмококка, гемофильной палочки, менингококка. Используются антибиотики из группы пенициллинов (амоксициллин). При развитии тромбозов применяются антикоагулянты (низкомолекулярные, нефракционированные гепарины).
- Гемотрансфузии. Так как анемия у пациентов с количественными патологиями гемоглобина всегда тяжелая, основу лечения составляют регулярные гемотрансфузии. Людям, страдающим качественными гемоглобинопатиями, переливания крови проводятся только при секвестрационных, гемолитических, а также апластических кризах.
- Борьба с перегрузкой железа и дефицитом фолатов. Для выведения избытка железа из организма используются хелатирующие средства (дефероксамин). Этот препарат обычно назначается вместе с аскорбиновой кислотой, так как она потенцирует хелатирующее действие дефероксамина. Из-за постоянного гемолиза у больных повышен расход фолатов, поэтому им показан длительный прием больших доз фолиевой кислоты.
Хирургическое лечение
Для ряда пациентов с выраженными признаками гемолиза эффективным лечением является спленэктомия – оперативное удаление селезенки. Другой хирургический вид лечения, позволяющий добиться полной ремиссии – аллотрансплантация гемопоэтических стволовых клеток. Однако этот метод применяется редко, только в очень тяжелых случаях, так как он сопряжен с высокой частотой летальных исходов. При холелитиазе выполняется холецистэктомия.
Экспериментальное лечение
В настоящее время ведутся клинические исследования по поиску лекарства для излечения гемоглобинопатий. Есть успешные результаты генной терапии СКА. Суть лечения заключается во введении в стволовые клетки пациента гена, кодирующего синтез нормальной бета-глобиновой цепи, с помощью обезвреженного лентивируса. Препарат называется LentiGlobin BB305. Его применение привело к улучшению показателей крови, что позволило отказаться от постоянной стандартной терапии. Также проводятся испытания данного препарата при бета-талассемии.
Прогноз и профилактика
Гемоглобинопатии являются тяжелыми заболеваниями с опасными жизнеугрожающими осложнениями. Пациенты с гомозиготной альфа-талассемией умирают еще до рождения в утробе матери. Больные бета-талассемией погибают до наступления пубертатного возраста от сердечной недостаточности. Люди, имеющие качественные гемоглобинопатии, при грамотном лечении могут прожить дольше 50 лет. Основная причина смерти – бактериальные инфекции, тромботические осложнения. При гетерозиготных формах заболевания в большинстве случаев продолжительность жизни не отличается от общей популяции.
Первичная профилактика проводится среди семей, имеющих высокий риск развития гемоглобинопатий. Она заключается в пренатальной диагностике с прерыванием беременности по медпоказаниям. Пациенты, страдающие качественными аномалиями гемоглобина, обязательно должны получить вакцину от гемофильной палочки, пневмококка, менингококка. Детям от 4 месяцев до 6 лет показано длительное применение пенициллиновых антибиотиков для профилактики инфекций. То же касается больных, перенесших спленэктомию.
Гемоглобинопатии. Причины, симптомы и прогноз при гемоглобинопатиях
Гемоглобинопатии – группа заболеваний, характеризующихся нарушением структуры гемоглобина или синтеза глобиновых цепей. Для первого вида самой распространённой является серповидно-клеточная анемия. Гемоглобинопатия количественного типа – это большая талассемия, или анемия Кули. В целом, аномалий гемоглобина насчитывается порядка 500 видов, большинство из которых не имеет клинических проявлений. Однако такие заболевания, как анемия, достаточно опасны и выражены специфическим гипоксическим симптомокомплексом.
Гемоглобинопатия – это наследственное заболевание. Поэтому первоочередные профилактические меры обычно направлены на перинатальную диагностику и медико-генетическое консультирование. Для многих видов гемоглобинопатий характерна распространенность в определённых географических границах (Африка, Западная Азия, Кавказ, Индия, Средиземноморье). Наибольшее распространение гемоглобинопатии получили в так называемом «малярийном поясе» – в районах с тропическим и субтропическим климатом.
Наследование этого заболевания влияет на клиническую картину или отсутствие таковой. Проявление унаследованной гемоглобинопатии не зависит ни от пола ребёнка, ни от пола родителя – носителя гена. Только когда оба родителя передают ребёнку ген гемоглобинопатии одного вида, проявляется ярко выраженная симптоматика. Получение признака аномалии гемоглобина от одного родителя может вообще никак не проявляться, оставаясь рецессивным.
Нередко гомозиготное наследование гемоглобинопатии приводит к гибели плода ещё до рождения. Если же ребёнок выживает, то он становится хронически больным с периодами обострений и относительного благополучия в течение всей жизни.
2. Основные симптомы заболевания
- бледность;
- утомляемость и слабость;
- увеличенная селезёнка;
- тёмная моча;
- желтушность кожи;
- низкая масса тела;
- специфическая форма черепа;
- увеличенный живот;
- низкий гемоглобин;
- ускоренное разрушение эритроцитов.
3. Диагностика болезни
Помимо визуального изучения клинической картины, для уточнения диагноза могут проводиться: общий анализ крови, электрофорез гемоглобина, проба на щелочную устойчивость эритроцитов.
Поскольку гемоглобинопатия неизлечима, больные могут лишь получать пожизненную симптоматическую медицинскую помощь и соблюдать ряд предписаний, которые снижают частоту и тяжесть обострений.
4. Лечение гемаглобинопатии
Лечение и поддерживающая терапия включают:
- препараты железа;
- фолиевую кислоту;
- диету, богатую белками и витаминами;
- глюкокортикостероиды;
- иммунодепрессанты;
- переливания крови и эритроцитарной массы;
- постельный режим в периоды обострений;
- удаление селезёнки;
- пересадку костного мозга.
Прогноз для больных гемоглобинопатией во многом зависит от типа патологии и степени её влияния на функции организма. Тяжёлые формы вызывают задержку развития и нередко гибель в детском возрасте. Особенно опасным фоном в периоды обострений становятся инфекции. В любом случае больные гемоглобинопатией постоянно находятся под медицинским наблюдением и требуют регулярного обследования и поддерживающих мер.
Заболевания Жалобы и симптомы- Высокая температура тела
- Иммунитет снижен
- Боли различной локализации
- Резкое снижение или набор веса
- Плохая работы желудка тракта
- Частые ОРЗ, ОРВИ
- Слабость, головокружение, недомогания
Если вы обнаружили у себя подобные симптомы, возможно, это сигнал заболевания, поэтому рекомендуем проконсультироваться с нашим специалистом.
Диагностика- ЭКГ (электрокардиограмма)
- Общий анализ крови
- Биохимический анализ крови
- Общий анализ мочи
- Пульсоксиметрия
- Консультация терапевта - от 1500 р.
- Биохимический анализ крови (стандартная, 10 показателей) - 2470 р.
- Биохимический анализ крови (расширенаяая, 14 показателей) - 3565 р.
- Общий анализ крови - 675 р.
- ЭКГ (электрокардиография) - 1500 р.
- Общий анализ мочи - 320 р.
- Пульсоксиметрия - 500 р.
Мы стараемся оперативно обновлять данные по ценам, но, во избежание недоразумений, просьба уточнять цены в клинике.
Данный прайс-лист не является офертой. Медицинские услуги предоставляются на основании договора.
Читайте также:
- Дизопирамид при аритмии сердца - показания, противопоказания
- Кровь при пищевых токсикоинфекциях. Гемограмма при вирусных кишечных инфекциях
- Диагностический амниоцентез при двойне. Лечебные вмешательства при многоплодной беременности
- Рефлекс на растяжение в клинике. Клонус мышечных волокон
- Протеинкиназы регулирующие вступление клетки в митоз