
Болезнь альцгеймера и прионы

Вверху - регуляция Циклофилином Б нормального сворачивания белка Пресенилина-1. Внизу патологическое нарушение этого процесса.
Изображение: Ehud Cohen et als. / The EMBO Journal
Международная команда ученых под руководством исследователей из Еврейского университета в Иерусалиме обнаружила, что в основе нескольких разных нейродегенеративных заболеваний, таких как болезнь Альцгеймера и прионные поражения мозга, лежит один общий механизм, связанный с появлением мутаций, которые мешают сворачиванию белковой цепи и взаимодействию с помогающими в этом процессе шаперонами — белками, главная функция которых состоит в восстановлении правильной третичной и четвертичной структуры других белков. Также ученые установили, что, вероятно, болезнь Альцгеймера является группой различных заболеваний, имеющих принципиально разные причины. Работа опубликована в The EMBO Journal.
Исследователи изучили паттерны мутаций, связанные со всеми известными нейродегенеративными заболеваниями. Выяснилось, что семейная болезнь Альцгеймера и некоторые наследственные прионные заболевания имеют очень схожий паттерн мутаций. Все они были связаны с заменой аминокислоты пролина — в одном случае в прионах, а в другом в белках Пресенилин-1 (семейство трансмембранных белков). Дополнительные эксперименты показали, что эта замена приводит к тому, что мутантный пресенилин не может взаимодействовать с другим белком — шапероном Циклофилином Б. Последний помогает правильному сворачиванию и приобретению необходимого пространственного строения полипептидной цепи у Пресенилинов-1.
Здесь важно также отметить, что пролин отличается от других аминокислот тем, что сильно изгибает белковую цепь. Замена пролина на любую другую аминокислоту сильно нарушает его конфигурацию. Недостаток Циклофилина Б или нарушения в процессе взаимодействия с ним при сворачивании, по мнению ученых, и лежит в основе обоих нейродегенеративных заболеваний.
Дополнительно исследователи обнаружили, что такая мутация сопровождает только ряд случаев болезни Альцгеймера. У многих других людей, страдающих схожими симптомами, ее нет, зато имеются другие мутации. Ученые считают, что, вероятно, болезнь Альцгеймера — это группа из нескольких заболеваний, с разными причинами, разным возрастом манифестации, но схожими симптомами. Именно поэтому какая-либо одна терапия не имеет устойчивого эффекта и не работает для всех пациентов одинаково. Важно отметить, что болезнь Альцгеймера может носить спорадический характер, и в этом случае она проявляется на седьмом десятке жизни пациентов или позже, а может быть семейной, тогда первые симптомы можно наблюдать уже на пятом десятке лет.
Пресенилины — семейство трансмембранных белков. Они кодируются двумя генами PSEN1 и PSEN2. Мутации гена PSEN1 ведут к синтезу бета-амилоида, и появлению амилоидных бляшек в мозге больных семейной болезнью Альцгеймера. Нейродегенеративные заболевания в настоящее время являются неизлечимыми. Их симптомы проистекают из дегенеративного развития и последующей смерти нервных клеток.
Как и пациенты с прионным заболеванием, больные Альцгеймером страдают от смертельной прогрессирующей формы деменции. Растет доказательство того, что агрегаты амилоида-β (Aβ) могут быть переданы аналогично прионам, по крайней мере в экстремальных экспериментальных условиях. Однако, в отличие от мышей, инфицированных прионами прионного белка (PrP), инокулированные Aβ не умирают. Таким образом, передача Aβ и PrP заметно отличается от неврологических эффектов, которые они вызывают у своих хозяев, причем разница не меньше, чем вопрос жизни и смерти. Это не просто академический нюанс, это различие между Aβ и PrP вызывает решающие вопросы того, что именно контролирует прионную токсичность и как прионная токсичность относится к инфекционности прионов.
В 1982 году Стэнли Прусинер ввел прионы в качестве самовоспроизводящихся форм прионного белка, которые накапливаются при некоторых трансмиссивных заболеваниях центральной нервной системы, таких как скрепи и болезнь Крейтцфельдта-Якоба1. Хотя прионы представляют собой новые инфекционные агенты, лишенные нуклеиновых кислот, кодируемых патогенами , их открытие опиралось на вековую парадигму, формализованную в постулаты Роберта Коха, для выявления патологических микробных агентов. Ключевая концепция в постулатах Коха заключается в том, что микроб, ответственный за данное заболевание, должен вызывать то же заболевание при инокуляции в восприимчивый хозяин. При прионном заболевании страдающий человек страдает от прогрессирующего ухудшения неврологической функции, которое завершается, неизбежно, в смерти. Путем систематического просеивания мозговых экстрактов из зараженных scrapie хомяков, Прусинер обнаружил, что самые смертоносные инокуляции содержат фибриллярные агрегаты протеолитического фрагмента прионного белка PrP27-30. Теперь мы знаем, что этот фрагмент получен из изоформы scrapie прионного белка PrPSc, агрегированного, альтернативно сложенного конформера клеточного прионного белка, PrPC.2
В 2000 году Лэри Уокер впервые продемонстрировала, что внутримозговые прививки мозговых экстрактов из мозговой ткани, содержащей амилоидный бляшко от пациентов с болезнью Альцгеймера, ускоряют осаждение амилоидных бляшек и β-амилоидоз у трансгенных мышей, экспрессирующих белки человека Aβ3. Ускорение β-амилоидоза инокулятами, содержащими Фибриллы Aβ, которые образуют амилоидные бляшки, были воспроизведены, по меньшей мере, в четырех других лабораториях с использованием инокулятов у людей, нескольких линий формирующих бляшку трансгенных мышей и, в последнее время, фибриллярных синтетических агрегатов Aβ и синтетических димеров Aβ.4-7
Прионы выполняют вышеуказанное определение, поскольку они были первоначально обнаружены как истинные инфекционные агенты с использованием микробиологических методов. Однако многие другие белки могут объединяться в геометрически расположенные структуры, которые могут вносить in vitro и in vivo-компартменты, содержащие исходный белок в мономерном растворимом состоянии.
К счастью, нет никаких признаков того, что такие процессы существуют для прионоидов Aβ. Тем не менее, демонстрация межличностной прозрачности будет способствовать повышению статуса таких агентов для добросовестных прионов, что, по-видимому, очень вероятно в случае амилоида AA.13.
До эпохи молекулярной генетики нейродегенеративные заболевания пожилых людей определялись типами несвязанных белков, которые накапливаются как нерастворимые отложения в мозге. Отложения состояли из высокоупорядоченных стеков β-листов, что является физическим определением амилоида. Каждый тип амилоида находится в характерном ядерном, цитоплазматическом или внеклеточном отделе. С появлением молекулярной генетики было обнаружено причинные гены, связанные со многими из этих нейродегенеративных заболеваний, и в удивительно большом числе случаев данный ген кодировал сам белок, содержащий амилоидные фибриллы, которые характеризовали невропатологию в этом заболевании.
Эти заболевания стали известны как расстройства, связанные с расстройством белков, поскольку каузальные гены кодировали несогласованные белки, включающие амилоидные поражения. В каждом случае несогласованные белки приобретают вторичную структуру-β-листы, которая отсутствует в нормальных условиях. Сопротивление белка в нейродегенеративных заболеваниях относится к условиям, в которых родительские белки берут новую вторичную структуру β-листа. Приобретение новой вторичной структуры отличает расстройства, связанные с расстройством белков, от других заболеваний, вызванных мутациями, которые изменяют конформацию белка, например, серповидноклеточную анемию.
За исключением PrPSc, нет экспериментальных доказательств того, что прионы или прионеиды в нейродегенеративных заболеваниях являются патогенными белками (звездообразными (*) белками], вызывающими неврологическое ухудшение, которое разрушает пациентов. Считается, что столетие нейрофибриллярные клубочки — внутриклеточные амилоидные включения, которые образуются, когда tau принимает новую структуру β-листа, — индуцируют гибель нейронов и ухудшают познание. Однако в 2005 году эта столетняя гипотеза была опровергнута, когда было показано, что уменьшение растворимого тау в модели нейродегенеративной мыши с нейрофибриллярными путаницами привело к прекращению потери нейронов и улучшению функции памяти, несмотря на поразительное наблюдение, что нейрофибриллярные клубочки сохраняли накопление, напоминающее прионеиды.14 Отрицательный случай для β-амилоидных бляшек, содержащих * белки, связан с несколькими линиями доказательств, включая отказ пациентов с болезнью Альцгеймера улучшить после иммунотерапии Aβ, которые, тем не менее, успешно удаляли амилоидные бляшки, 15 и способность иммунотерапии для устранения дефицита у мышей без изменения нагрузки на бляшку.16,17 По сравнению с белками, включающими амилоидные поражения, прионы и прионеиды, относительно мало известно о белках и их механизмах действия.
Безусловно, наиболее понятным среди этих расстройств является семейная атаксия типа 1. Атаксин-1 вызывает семейную атаксию типа 1, когда посторонние последовательности тринуклеотидных повторов расширяют существующий полиглутаминовый тракт с образованием PolyQ / ataxin-1.18. Пациенты и мыши, экспрессирующие PolyQ / ataxin-1, развивают ядерные включений и прогрессирующей атаксии. Агрегаты PolyQ являются прионоидами, поскольку применение агрегатов PolyQ к культивируемым нейронам зарождает образование внутриклеточного полиомиелита амилоида.19 У мышей, несущих разреженный расширенный тракт PolyQ, тяжесть неврологических аномалий и нейродегенераций обратно пропорциональна числу включений, 20 и отмена включений посредством мутации лигида ubiquitin, которая способствует агрегации несогласованных белков, ускоряет заболевание.21 Эти исследования показывают, что элиминация включений не будет препятствовать атаксии в семейной атаксии типа 1.
Вывод о том, что удаление включений не вылечило семейную атаксию типа 1, побудило Гарри Орра и Худу Зогби искать патогенную форму PolyQ / ataxin-1, вызывая неврологические аномалии в расстройстве. Обнаружение механизма, с помощью которого PolyQ / ataxin-1 повреждает нейроны, возникло из понимания нормальной физиологической роли атаксина-1, являющегося ядерным белком. Патогенная форма PolyQ / ataxin-1 не является неправильной формой атаксина-1; он не содержит новых вторичных структур, нет β-листов, которые обычно не присутствуют в мозге. Его патологические эффекты возникают из-за изменений в его аффинности связывания с его нормальными ядерными партнерами, транскрипционного регулятора Capicua и регулятора RNA-сплайсинга RMB17,22, что приводит к изменениям транскриптома, которые, по-видимому, влияют на нейронную функцию и жизнеспособность. Таким образом, патогенная форма PolyQ / ataxin-1 не является прионоидом; он не является ни ложным белком, ни растворимым агрегатом исходного белка. Это может оказаться глубоко важным уроком для всей области исследований нейродегенеративных заболеваний.
Оба пациента с прионным заболеванием и болезнью Альцгеймера страдают от смертельных прогрессирующих форм деменции. Однако, в то время как мыши, зараженные PrP-прионами, умирают, у тех, кто инокулируется с прионами Aβ, нет. Только две гипотезы могут объяснить резкий контраст между коэффициентами летальности, вызванными привитиями PrP и Aβ у мышей.
Агрегаты являются патогенными, но разные агрегаты оказывают влияние на различные клеточные пути. Например, патогенный путь для агрегатов Аβ у людей, отличный от таковых прионов PrP, может отсутствовать у мышей.
Агрегаты не всегда являются патогенными; скорее, варианты исходных белков (* белков) вызывают клеточную дисфункцию, которая приводит к неврологическому заболеванию (рис.1). Эти патогенные варианты не обязательно должны быть неправильными или агрегированными формами исходных белков.
Недавние достижения в нашем понимании нейротоксичности PrP и Aβ благоприятствуют гипотезе 2, как обсуждается ниже.
При прионном заболевании катастрофическая дисфункция мозга связана с глобальным снижением производства белка, вызванным дисрегуляцией eIF2a, фактором инициации трансляции млекопитающих23. Это увлекательное открытие, по-видимому, является механизмом, с помощью которого прионы PrP в конечном итоге вызывают нейротоксичность.
Однако eIF2a локализуется внутри цитозоля, тогда как инфекционные прионы являются внеклеточными. Таким образом, нам все еще остается интересно, как прионы, содержащие патологически агрегированный PrPSc, могут оказывать действие, происходящее из внеклеточной среды, сгибание белков дерьма в эндоплазматическом ретикулуме, индуцировать неожиданно энергичный развернутый белковый ответ и в конечном итоге гасить цитозольный перенос белков. Трудно не заключить, что подавление eIF2a, вероятно, представляет собой нисходящий эффектор патогенного каскада, который инициируется молекулярно и топологически отдаленными событиями.
Рисунок 2. Клеточный прионный белок абсолютно необходим для токсичности инфекционных прионов (А) 39, что подразумевает, что PrPSc оказывает нейротоксичность путем стыковки с PrPC (B). Эта токсичность также может быть вызвана вариантами PrP, встречающимися естественным образом, такими как PrP, несущими сверхматериальные октапептидные повторы (C), или экспериментально сконструированные токсичные варианты, такие как версии PrP, содержащие делеции шарнирной области (D). Недавно было обнаружено, что прионная инфекция приводит к цепочке событий, которая в конечном счете утоляет перенос белка 23, но остается неизвестным, использует ли токсичность, вызванную мутантами PrP (Панели C и D), тот же путь.
Наше понимание нейротоксичности Aβ отстает от нейротоксичности PrP, потому что животные модели, которые пересказывают все аспекты болезни Альцгеймера, которые необходимы для анализа человеческой значимости патологического Aβ, не существуют. У людей патологическая трансформация Aβ инициирует процесс, который включает накопление амилоидных бляшек и часто приводит к фатальному нейродегенеративному состоянию. У мышей образование патологического Аβ может индуцировать осаждение амилоидных бляшек, что отражает присутствие прионоидов Aβ. Удивительно, что накопление прионоидов Aβ у подавляющего большинства мышей не приводит к открытой нейродегенерации, за исключением непосредственной близости от амилоидных бляшек.28-30 У мышей, лишенных синтазы оксида азота 2, накопление априонидов Aβ связано с нейронов в CA3, но не подполе CA1 hippocampal 31, но у людей CA3 сохраняется, а CA1 — нет, 32 ставит под сомнение значимость этой формы смерти нейронов. Независимо от того, является ли смерть нейронов в этой модели из-за априонидов Aβ, также неизвестно. Фатальность, когда она присутствует, является зависимой от деформации и может возникать в отсутствие образования амилоидов, что указывает на существование нейротоксического вида, который не обязательно может быть сопоставим с самораспространяющимися видами.33 Биохимическая идентичность и клеточные эффекты этого форма Aβ остаются неизвестными.
Два типа когнитивной дисфункции развиваются у мышей с образованием Aβ. Один тип встречается у мышей с агрессивным отложением амилоидов — чрезмерное количество прионоидов Аβ, в которых накопленные амилоидные бляшки и их окружающая цитопатология действуют как гигантское космическое поражение в мозге.28 Никакие нейроны не остаются в сердцевинах бляшек и извилистые , дистрофические нейриты и дендриты, частично оголенные шипами, находятся в 50-микронном гало, окружающем ядра (рассмотренные Ashe и Zahs34). Поэтому неудивительно, что за определенным порогом познание изменяется обратно пропорционально нагрузке на бляшку. Однако плотность амилоидных бляшек, необходимых для получения этого эффекта, редко достигается при болезни Альцгеймера, что может объяснить, почему связь между бляшками и потерей или познаниями в нейронах в лучшем случае незначительна.35,36 Чрезмерное накопление прионоидов Aβ у мышей может привести к в когнитивных дефицитах, которые связаны с пространственно-занимающими эффектами амилоидных бляшек, но это отличается от механизма, с помощью которого большая часть нейрональной дисфункции и нейродегенерации происходит у людей с β-амилоидозом или болезнью Альцгеймера.
Другой тип когнитивной дисфункции происходит независимо от бляшек или потери нейронов и, по-видимому, обусловлен нефибриллярной сборкой Aβ 56 см-1 Aβ, обозначенной Aβ * 56.37 Aβ * 56, предполагаемым додекамером, который, скорее всего, образован кластеризацией четырех Aβ-тримеров , Изоляты фибриллярного Aβ в головном мозге не содержат Aβ * 56 (Liu P и Ashe K, неопубликованные данные). Маловероятно, что Aβ * 56 приобретает новую структуру β-листа, поскольку композиционная единица Aβ-тримеры присутствует даже у молодых мышей.37 Отсутствие новой структуры β-листа в Aβ * 56 утверждает, что она является прионеодом, хотя формальная демонстрация потребует экспериментов по передаче. Aβ * 56 не вызывает явной нейродегенерации и все же изменяет познание, нарушая долговременную синаптическую пластичность еще неизвестным механизмом.38 У людей Aβ * 56 можно измерить в CSF, где он увеличивается с нормальным старением и коррелирует умеренно сильно с белком, связывающим микротрубочки тау (М. Ханкоко, М. Грант, А. Валлин, К. Бленноу и К. Эше, неопубликованные данные). Тау высвобождается в CSF, в процессе, который остается неясным, когда нейроны неисправны. CSF tau слабо коррелирует или вообще отсутствует с CSF Aβ1-42, который отражает β-амилоидоз и априониды Aβ.
Хотя было заманчиво постулировать, что Aβ * 56 вызывает последовательность событий, которая приводит к переходу от бессимптомного старения к умеренным когнитивным нарушениям или болезни Альцгеймера, что совпадает с началом открытой нейродегенерации и потерей нейрона, это предсказание не подтвердилось в большое продольное исследование (Handoko M, Grant M, Petersen R и Ashe K, неопубликованные данные). Таким образом, Aβ * 56 нарушает функцию нейронов у мышей и связывается с нейронной неисправностью у людей, но недостаточно для индукции явной нейродегенерации у обоих видов.
В отсутствие животных моделей, содержащих исключительно мутаций, связанных с болезнью Альцгеймера, которые проявляют полный спектр заболеваний, начиная с тонкой дисфункции нейронов и заканчивая фатальной когнитивной опустошением, вопрос о том, требует ли асимптоматический β-амилоидоз Aβ * 56 в полной мере Продуманная болезнь Альцгеймера не может быть рассмотрена экспериментально. Возможно, что одна или несколько неприноидных форм Aβ запускают нейронную дисфункцию и нейродегенерацию при болезни Альцгеймера. Обнаружение этих патогенных форм будет зависеть от создания высококачественных модельных систем болезни Альцгеймера.
При доброкачественных прионных заболеваниях очень большой объем данных связывает агрегированную форму PrP, PrPSc, как с инфекцией прионов, так и прионной нейротоксичностью. Тем не менее, неинфекционные, но нейротоксичные варианты PrP встречаются естественным образом, и более такие варианты были построены экспериментально, что указывает на то, что фенотипическое выражение, типичное для прионных заболеваний, может быть вызвано событиями, происходящими после прионной инфекции. Существует мало свидетельств того, что мыши или люди связывают неврологические эффекты Aβ с зародышеобразующими формами этого белка, в то время как новые данные указывают на конкретную не зародышевую форму Aβ, Aβ * 56, которая вызывает некоторые неврологические признаки болезни. Однако Aβ * 56 не является достаточным для индуцирования неумолимого неврологического ухудшения, которое характеризует болезнь Альцгеймера, указывая на то, что другие критические факторы или формы Aβ работают в сотрудничестве с Aβ * 56 для разрушения мозга. Лечение приона и болезни Альцгеймера будет зависеть от более глубокого понимания патогенных форм PrP и Aβ, которые вызывают дисфункцию мозга, лежащую в основе этих смертельных заболеваний.
Возможные конфликты интересов выявлены не были.
Болезнь Альцгеймера является формой деменции, группы нейродегенеративных заболеваний, вызванных токсическими скоплениями дефектных белков. Известно, что прионы, которые являются "битыми" версиями белков, вызывают некоторые редкие формы деменции. Эти белки содержат ненормально свернутый белок, который заставляет нормально свернутые белки ненормально складываться и превращаться в аномальную форму приона. Эти новые аномально свернутые белки затем вызывают те же изменения в других белках и в конечном итоге образуют бляшки, которые препятствуют синаптической передаче в мозге (синапсы представляют собой химические соединения между соседними нервными клетками). Получающиеся в результате бляшки приводят к повреждению и гибели нервных клеток, что в конечном итоге приводит к нейродегенеративным заболеваниям, таким как деменция. Пациенты с прионными заболеваниями имеют широкий спектр симптомов: от нарушения работы мозга до изменений личности и трудностей с подвижностью.

Первый прионный белок был открыт в 1980-х годах Стэнли Прусинером, доктором медицины, который был старшим автором нового исследования. В его новой работе исследователи объединили два лабораторных теста для измерения уровня прионов в образцах тканей человека. Методы включают новую систему обнаружения Aβ, созданную в лаборатории Прусинера, и анализ на тау-прионы. Эти клеточные анализы могут измерять уровни инфекционных прионов за три дня, что значительно сокращает время, необходимое для проведения такого рода исследований и анализа. Они позволили исследователям быстро рассчитать количество прионов tau и Aβ в образцах мозга после смерти. Для исследования было проанализировано более 100 образцов вскрытой мозговой ткани, взятых у пациентов, умерших от болезни Альцгеймера, среди других типов нейродегенеративных состояний в США, Европе и Азии.
Результаты показали, что в 75 образцах ткани головного мозга людей, которые умерли от болезни Альцгеймера, Aβ и тау-приона, активность была высокой. 11 образцов от людей, которые умерли от церебральной амилоидной ангиопатии (CAA), содержали только прионы Aβ. 10 образцов лобно-височной долевой дегенерации с тау-патологией (FTLD) содержали только тау-прионы.
Читайте также: ВОЗ: головоломки и таблетки не защитят от деменции
Было также обнаружено, что прионные формы тау и Aβ наиболее распространены у пациентов, которые унаследовали заболевание и умерли от него в более молодом возрасте. В резком контрасте с обычными паттернами, наблюдаемыми на пути болезни у пожилых пациентов, было также обнаружено, что прионные формы тау значительно снижались с увеличением возраста, а не увеличивались в более старшем возрасте.
Была обнаружена сильная корреляция между более молодым возрастом и более высоким уровнем тау-прионов. В среднем, у пациентов, которые скончались в возрасте 40 лет, уровни тау-приона в 32 раза выше, чем у пациентов, которые скончались в возрасте 90 лет. Соавтор исследования химик Уильям ДеГрадо принимал участие в разработке и анализе нового исследования. Он описал анализы как-то, что "изменит правила игры", сказав, что:
"Ранее исследование болезни Альцгеймера ступорилось, когда рассматривало сопутствующее повреждение в виде неправильно сложенных мертвых белков, которые образуют бляшки и треугольники. Теперь выясняется, что именно прионная активность связана с болезнью, а не количество бляшек во время вскрытия".
Фото превью: Shutterstock
Встройте "Правду.Ру" в свой информационный поток, если хотите получать оперативные комментарии и новости:
Подпишитесь на наш канал в Яндекс.Дзен или в Яндекс.Чат
Добавьте "Правду.Ру" в свои источники в Яндекс.Новости или News.Google
Также будем рады вам в наших сообществах во ВКонтакте, Фейсбуке, Твиттере, Одноклассниках.

Рубрика: Медицина
Дата публикации: 02.09.2019 2019-09-02
Статья просмотрена: 33 раза
Болезнь Альцгеймера (БА) является наиболее распространенным нейродегенеративным заболеванием у человека и представляет собой значительную проблему для системы здравоохранения. БА является наиболее распространенной причиной деменции у людей. Частота развития БА зависит от возраста: у
1 из 9 человек в возрасте 65 лет и старше развивается БА, в то время как
1 из 3 человек в возрасте 85 лет и старше получает это заболевание [1].
Поражающий фактор БА характеризуется церебральными амилоидными отложениями, образованными из амилоидного β-пептида (Aβ), потерей нейронов и внутриклеточными отложениями — нейрофибриллярными клубками (NFT), агрегацией гиперфосфорилированных форм белка, ассоциированных с микротрубочками tau (τ) [2].
Изучение этиологии возникновения БА привело учёных к генетике. Генетические мутации, вызывающие БА, были обнаружены в гене APP (белок-предшественник амилоида). Современное открытие, что Aβ был нормальным продуктом метаболизма APP на протяжении всей жизни, позволило ученым быстро установить биохимические аномалии, вызванные мутациями APP. Генетический анализ разнообразных родственных семей болезни Альцгеймера указывает на биосинтез амилоидного β-пептида, генерируемого опосредованным секретазой эндопротеолизом APP, который является общим знаменателем в наследственных формах заболевания [3]. Считается, что мутации в пресенилинах 1 и 2 усиливают расщепление АРР в сайте γ-секретазы [4].
Наконец, установлено, что аллель ε4 гена ApoE, который коррелирует с повышенной восприимчивостью к позднему началу БА [5], усиливает образование зрелых бляшек у некоторых трансгенных мышей APP [6]. Кроме того, мутации АРР, внутренние по отношению к последовательности Аβ, усиливают самоагрегацию Аβ в амилоидные фибриллы [7]. Эти генетические данные указывают на то, что повышенный биогенез или накопление Аβ, вероятно, является критическим патогенным событием при всех формах БА. Этот вывод находит параллель в исследованиях, указывающих на то, что Аβ является нейротоксичным [8].
Одной из основных причин нейродегенеративных заболеваний является инфекционный белок — прион, получивший обозначение PrP SC [9]. Инфекционный прионный белок PrP SC образуется в результате конформационных изменений третичной и четвертичной структуры нормального клеточного белка PrP C . Происходит соединение молекул PrP SC и PrP C и образование 2-х молекул PrP SC , что обеспечивает экспоненциальный рост количества молекул PrP SC [10].
Белки могут подвергаться воздействию внутренних и внешних сил. Поскольку эти силы чередуют конформацию белка, биологическая активность белка снижается. Однако вновь синтезированные белки могут неправильно складываться. В этом случае белки имеют сильную тенденцию к агрегации [11]. Белковые агрегаты оказывают токсическое действие при накоплении в клетке определенного количества. Накопление аномальных белков приводит к прогрессирующей потере структуры и функции нейронов, включая гибель нейронов [12].
Все чаще предполагается, что прионы при амилоидных заболеваниях являются промежуточными звеньями на пути образования амилоидных структур [13]. Как известно, взаимодействие белка PrP С и Aβ проходит так называемую жидкую фазу. В жидкой фазе α-спиральный треонин становится раскрученным. На поверхности клетки Prp-лизиновые остатки взаимодействуют с олигомерами Aβ (Aβ0), после чего образуется гидрогель, состоящий из неподвижного Aβ0 и ограниченно подвижного PrP С . Aβ0/PrP-гидрогель имеет хорошую стоихометрию и диссоциирует с избыточным Aβ0. Aβ0/PrP-гидрогель захватывает сигнал-проводящую mGluR5 на плазматической мембране, что приводит к потере синапсов [14].
Считается, что у пациентов с БА отложения Aβ постепенно распространяются по всему мозгу, что обусловливает механизм распространения на основе прионов [15]. Агрегация и отложение амилоидных пептидов являются центральными событиями в патогенезе БА. Инокуляция гомогенатов головного мозга, содержащих агрегаты Aβ, в восприимчивых трансгенных мышей ускоряет отложение Aβ, подтверждая, что агрегаты Aβ способны к самораспространению и, следовательно, могут быть прионами [16].
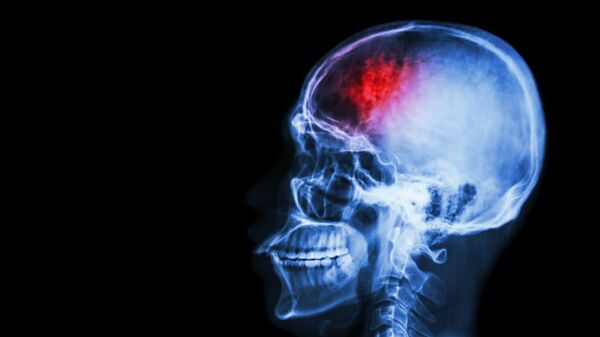
МОСКВА, 16 фев — РИА Новости. Врачи выяснили, что белки-прионы, вызывающие болезнь Альцгеймера, могут передаваться не только при переливании крови или инъекциях гормона роста, но и во время обычных нейрохирургических операций, говорится в статье, опубликованной в журнале Acta Neuropathologica.
В сентябре 2015 года Бранднер и его коллеги, изучавшие мозг людей, погибших от коровьего бешенства, обнаружили, что болезнь Альцгеймера может передаваться от одного человека к другому в результате заражения нервной ткани чужеродными прионами.
Прионы представляют собой клубки спутанных и неправильно собранных белковых молекул, которые накапливаются в нервных клетках больных людей. Как оказалось, они способны стимулировать появление бляшек бета-амилоида у здоровых людей, если попадают в их кровь и мозг.
Это впервые поставило медиков перед тем, что болезнь Альцгеймера может быть заразной. Переносчиком и источником прионов, как обнаружили ученые еще в прошлом году, служат некоторые болезнетворные бактерии, а также зараженная кровь от доноров, страдающих от этой болезни.
Бранднер и его коллеги нашли новые доказательства заразности болезни Альцгеймера, проанализировав архивы Национального неврологического госпиталя Великобритании, где хранятся записи о случаях заболевания за последние несколько десятилетий.

Ученых интересовали сравнительно редкие и необычные операции, во время которых врачи пытались ликвидировать кровотечения в мозге, вызванные накоплением бляшек бета-амилоида и других прионов. Подобные проблемы, как отмечают биологи, чаще встречаются у пожилых людей с болезнью Альцгеймера и другими нейродегенеративными заболеваниями, а для молодых пациентов они крайне нехарактерны.
Руководствуясь этой идеей, ученые попытались найти в архиве заметки об операциях, где пациентами были не пенсионеры, а довольно молодые люди, непонятным образом заполучившие болезнь Альцгеймера. В общей сложности исследователям удалось найти четыре таких случая — двух молодых людей и одну женщину в возрасте от 30 до 40 лет и еще одну женщину 57 лет.
Все они перенесли сложные операции по удалению доброкачественных опухолей мозга, костей черепа и позвоночника примерно за 15-20 лет до появления первых кровотечений. Как показал статистический анализ, это совпадение не было случайным — они заразились клубками бета-амилоида в результате контакта с хирургическими инструментами.
"Пока нельзя точно говорить, что болезнь Альцгеймера заразна, — эти люди, несмотря на наличие бляшек бета-амилоида в их мозге, не страдали от других ее проявлений, в том числе аномально высоких уровней тау-белка. С другой стороны, все это указывает на то, что хирургам стоит радикально поменять способ стерилизации инструментов при подобных операциях", — заключает Бранднер.
Читайте также: