
Что такое невропатия дюшена

Мышечная дистрофия, или миопатия Дюшенна, — тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера, но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Этиология нарушения
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.

Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.

Мышечная дистрофия Дюшенна никогда не протекает в легкой степени, всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:

На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, — это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.

Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.

Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:

Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.

В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса. Запрещено употребление алкоголя, кофеина и крепкого чая.
Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.

Мышечные дистрофии – это группа заболеваний не воспалительного характера, характеризующиеся прогрессивным течением без патологических изменений центральной и периферической нервной системы.
- Что это такое?
- Этиология, кто наследует и почему
- Симптомы мышечной дистрофии
- Формы миопатии
- Методы диагностики
- Лечение заболевания
- Последствия и осложнения
- Прогноз на выздоровление и жизнь
- Что нужно запомнить?
Что это такое?
Первые публикации о миопатиях датируются 1830 годом. В 1852 году Мерион сообщил о случае в семье с четырьмя сыновьями, у которых отмечались признаки двигательных расстройств, не связанных с поражением нервной системы. Он предположил, что заболевание наследуется от матери к ребенку.
Под миопатией или миодистрофией Дюшенна (также ее называют дистрофией или миопатией Дюшена-Беккера) понимают заболевание генетической природы, связанное с мутацией гена, кодирующего синтез белка дистрофина.
Дистрофин входит в состав мембран мышечных клеток, без него мышцы крайне уязвимы и подвергаются некрозу и дегенерации. Экспрессия, то есть реализация функции, происходит в гладкой и скелетной мышечной ткани, а также в миокарде.
Миопатия Дюшенна поражает одного из 3500 новорожденных детей. Несмотря на тот факт, что заболевание названо именем Дюшенна, одним из первых врачей, описавших данную патологию, был Говерс.
Гийом Дюшенн был французским неврологом, который использовал электростимуляцию в лечении неврологических расстройств. В 1868 году Дюшенн описал 13 пациентов с заболеванием, которое сопровождалось прогрессирующей мышечной слабостью, и назвал ее паралитической псевдогипертрофической мышечной дистрофией. Также он определил диагностические критерии миодистрофии, которые используются и сегодня.
Этиология, кто наследует и почему
По международной классификации болезней МКБ-10 мышечная дистрофия Дюшенна-Беккера имеет код G71.
Одна треть случаев заболевания связана со спонтанно возникшей мутацией, в то время как на остальные случаи патологии приходится наследование Х-хромосомы, несущей патологический ген.
Случаи гонадного мозаицизма связаны с 20% случаев миодистрофии Дюшенна. Средняя продолжительность жизни менее 20 лет. Повышение уровня фермента креатинфосфокиназы (КФК) обнаруживают у 2/3 женщин – носителей патологического гена. Большинство из них не имеют никаких проявлений заболевания. Миопатия почти всегда поражает мальчиков, поскольку заболевание сцеплено с Х-хромосомой, тип наследования – рецессивный. Локализация гена, кодирующего синтез дистрофина, находится в локусе Xq21. Синтез белка кодируется одним из самых больших известных генов. Он занимает около 2% ДНК Х-хромосомы.
Симптомы мышечной дистрофии
Первые симптомы появляются в возрасте от трех до семи лет. Обычно родители замечают раскачивающуюся походку и гиперлордоз. Существует несколько основных критериев, которые позволяют предположить миопатию Дюшенна. К ним относятся:
- Мышечная слабость, которая появляется неожиданно и начинается с нижних конечностей.
- Гиперлордоз – выраженный изгиб позвоночника кпереди, что особенно проявляется при ходьбе.
- Гипертрофия ослабленных мышц.
- Слабый ответ мышц на электрическую стимуляцию на более поздних стадиях заболевания.
С течением времени все указанные симптомы прогрессируют. Несмотря на поражение мышечных структур, нарушений функции мочевого пузыря и кишечника не отмечается. К двенадцати годам жизни большинство пациентов не могут ходить самостоятельно, и находится в инвалидном кресле. Миопатия Беккера очень сходна с дистрофией Дюшенна, так как поражается один и тот же локус Х-хромосомы, в результате чего страдает синтез дистрофина. Отличием миопатии Беккера является начало заболевания, как правило, после трех лет жизни или даже в подростковом возрасте.
Миодистрофия Дюшенна – патология, которая затрагивает не только скелетную мускулатуру. Дистрофин также содержится в миокарде, тканях мозга и гладкомышечной мускулатуре. Поздняя стадия заболевания ассоциируется с тяжелой сердечной недостаточностью и дыхательными нарушениями – главными причинами смерти пациентов.
Формы миопатии
В течении заболевания различают пять стадий. Первая стадия (доклинических проявлений) не характеризуется широким спектром симптомов, обычно у пациентов отмечается лишь повышение уровня КФК в сыворотке крови.
Вторая стадия (ранних признаков) включает следующие симптомы:
- Раскачивающаяся походка – впервые появляется от 2-х до 6-ти лет и часто является первым симптомом, который замечают родители.
- Прогрессирующая слабость мускулатуры нижних конечностей с дальнейшим присоединением слабости мышц шеи, плечевого пояса и рук.
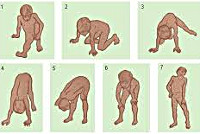
- Наличие симптома Говерса – при попытке встать с пола, пациент упирается на колени и руки (рисунок 1). Появление симптома связано с выраженной слабостью мышц спины и конечностей.

Рисунок 2. Симптом Говерса – пациент поднимается с пола, опираясь на колени и руки.
Третья стадия (прогрессирующих симптомов) характеризуется появлением значительных сложностей во время ходьбы и развивается, когда возраст ребенка составляет около восьми лет. Пациенту становится сложно подниматься по ступеням, появляется одышка, сложно встать с пола. По утрам могут беспокоить головные боли, в ночное время наблюдается затрудненное дыхание. Четвертая и пятая стадии являются наиболее тяжелыми, так как пациент теряет возможность не только самостоятельно передвигаться, но и испытывает трудности с дыханием. При четвертой стадии пациент еще может удерживать осанку, однако делать это становится все труднее, развивается выраженный сколиоз.
Пятая стадия – терминальная. Пациент не может ходить и, как правило, находится в инвалидном кресле. Чаще всего, смерть наступает от дыхательной и сердечно-сосудистой недостаточности в возрасте до 20-ти лет.
Методы диагностики
Ребенок с миопатией Дюшенна-Беккера до 2-3-х лет жизни может ничем не отличаться от других детей. Тем не менее, существует ряд признаков, на которые стоит обратить внимание.
Важно! Во время проведения лабораторных исследований может быть повышен уровень печеночных ферментов: аланин- и аспартатаминотрансферазы, креатинкиназы и гамма-глутаматтрансферазы.
В ряде исследований миопатий было отмечено, что задержка речи и моторных навыков отмечалась чаще у детей с дефектом дистрофина. Уровень IQ может быть меньше на одно стандартное отклонение по сравнению со среднестатистическим значением в популяции.
У 30% детей с мутациями гена, кодирующего синтез дистрофина, отмечались трудности с обучением и приобретением новых навыков, обсессивно-компульсивные расстройства, синдром дефицита внимания, задержка умственного развития. Детям с дистрофиями Дюшенна и Беккера сложнее даются вербальные навыки.
- Исследование уровня КФК, которая превышает референсные значения в 50-100 раз.
- Молекулярную диагностику – исследование гена, локализованного в локусе Xq21 и ответственного за синтез дистрофина, позволяет подтвердить или опровергнуть диагноз.
Если у пациента наблюдается сочетание высоких показателей КФК и мышечная слабость, это с высокой вероятностью позволяет заподозрить миопатию Дюшенна-Беккера.
Из инструментальных методов используют:
- обзорную рентгенографию грудной клетки. Исследование позволяет сделать заключение, насколько выражен сколиоз;
- электромиографию: метод применяют с целью дифференциальной диагностики со спинальной мышечной атрофией;
- электрокардиография – с целью выявления синусовых аритмий;
- ультразвуковое исследование сердца – нередко определяет малые размеры желудочков сердца и более длинную диастолу;
- холтеровское мониторирование определяет наличие пароксизмальных аритмий.
Если молекулярная диагностика не выявила мутации гена дистрофина, рекомендуется провести биопсию мышечной ткани. Характерные гистологические изменения приведены ниже:
- мышечные волокна с выраженным дегенеративным процессом и некрозом;
- пролиферация соединительной ткани;
- появление жировой ткани в значительном количестве.
Также проводится анализ белка дистрофина, выделенного из мышцы, с определением его молекулярной массы.
Лечение заболевания
На сегодняшний день лечения миодистрофии Дюшенна-Беккера не существует. Терапевтическая тактика основана на поддерживающем медикаментозном лечении (в основном, для сохранения сердечной функции), исследованиях в области генной инженерии и экспериментальном клеточном лечении.
При прогрессировании сколиоза и контрактур суставов возможно применение паллиативного хирургического вмешательства. По мере прогрессирования мышечной слабости, следует обеспечить пациенту максимальный комфорт.
Применение преднизолона активно обсуждается в профессиональных медицинских сообществах. Известно, что спустя один месяц от начала применения наблюдается незначительное улучшение общего состояния пациента и может сохраняться до трех лет без ухудшения. В то же время, при отказе от преднизолона начинается прогресс заболевания.
Последствия и осложнения
Главными осложнениями, с которыми сталкиваются пациенты, являются:
- Прогрессирующая мышечная слабость.
- Дилатационная кардиомиопатия.
- Дыхательные нарушения, связанные с дисфункцией диафрагмы.
- Контрактуры суставов.
- Сколиоз.
- Дисфагия.
- Запоры.
К осложнениям также относится остеопороз и высокий риск переломов. По некоторым данным возможно использование препаратов кальция и витамина Д для увеличения плотности костной ткани.
Прогноз на выздоровление и жизнь
К сожалению, прогноз неблагоприятный, так как смертность при данной патологии составляет 100%. Если в семейной истории встречается близкая родственница, которая является носителем патологического гена, у детей мужского пола выше риск развития кардиомиопатии с прогрессирующей сердечной недостаточностью в возрасте от 20 до 40 лет. Проводятся исследования, связанные с применением стволовых клеток для лечения миодистрофий.
Что нужно запомнить?
- Миопатия Дюшенна-Беккера – прогрессирующее заболевание, связанное с аномальным синтезом белка дистрофина, принимающего непосредственное участие в мышечных сокращениях. Синтез дефектного белка приводит к дегенеративным и некротическим процессам в мышцах.
- Ранними признаками болезни считаются появление мышечной слабости в нижних конечностях, раскачивающейся, нетипичной походки. К лабораторным признакам относится многократное повышение уровня КФК.
- Этиология заболевания – генетическая, связанная с мутацией гена в локусе Тип наследования-рецессивный, мутация может передаваться от матери, чья Х-хромосома несет патологический ген, а может возникать спонтанно.
- Существует пять стадий миопатии. Четвертая и пятая стадии являются терминальными.
- К ранним методам диагностики относится определение уровня КФК, который может многократно превышать норму и являться ранним маркером наследственной миодистрофии Дюшенна-Беккера задолго до появления мышечной слабости и других заметных симптомов. К более точным методам относится молекулярная диагностика с исследованием патологического гена.
- Специфическое лечение не разработано. Мероприятия по уходу за пациентом сводятся к поддержанию жизнеобеспечения.
- Основными осложнениями являются утрата способности ходить, тяжелые дыхательные расстройства и сердечно-сосудистая недостаточность.
- Прогноз неблагоприятный, летальность составляет 100%.
Литература
- Bushby K, Straub V. Nonmolecular treatment for muscular dystrophies. Curr Opin Neurol. 2005 Oct. 18(5):511-8.
- Moxley RT 3rd, Ashwal S, Pandya S, Connolly A, Florence J, Mathews K, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005 Jan 11. 64(1):13-20.
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991 Sep 20. 66(6):1121-31.
- Ozawa E, Noguchi S, Mizuno Y, et al. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve. 1998 Apr. 21(4):421-38.
- Darke J, Bushby K, Le Couteur A, McConachie H. Survey of behaviour problems in children with neuromuscular diseases. Eur J Paediatr Neurol. 2006 May. 10(3):129-34.
- Mendell JR, Shilling C, Leslie ND et al. Evidence based path to newborn screening for Duchenne Muscular Dystrophy. Ann Neurology. 2012. 71:304–313.
- Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR. Gene therapy for duchenne muscular dystrophy: expectations and challenges. Arch Neurol. 2007 Sep. 64(9):1236-41.
- Merlini L, Gennari M, Malaspina E et al. Early corticosteroid treatment in 4 duchenne muscular dystrophy patients: 14-year follow-up. Muscle Nerve. 2012 Jun. 45(6):796-802.
- American Academy of Pediatrics Section on Cardiology and Cardiac Surgery. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005 Dec. 116(6):1569-73.
- Colan SD. Evolving therapeutic strategies for dystrophinopathies: potential for conflict between cardiac and skeletal needs. Circulation. 2005 Nov 1. 112(18):2756-8.

Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
- Причины мышечной дистрофии Дюшенна
- Симптомы мышечной дистрофии Дюшенна
- Диагностика мышечной дистрофии Дюшенна
- Лечение мышечной дистрофии Дюшенна
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины мышечной дистрофии Дюшенна
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы мышечной дистрофии Дюшенна
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика мышечной дистрофии Дюшенна
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК. При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
Лечение мышечной дистрофии Дюшенна
Терапия, применяемая в клинической практике, пока включает лишь необходимые симптоматические мероприятия. Для улучшения метаболизма мышечной ткани возможно назначение анаболических стероидов (метандиенона, нандролона деканоата), АТФ, актопротекторов (этилтиобензимидазола); для облегчения нервно-мышечной передачи — неостигмина. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия.
Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Поиск эффективных способов лечения дистрофии Дюшенна — задача, над решением которой трудятся сегодня специалисты в области неврологии, биохимии, генной инженерии. Из перспективных разработок в этом направлении можно выделить лечение стволовыми клетками, активацию гена утрофина, являющегося наиболее близким аналогом дистрофина, технологию пропуска экзонов.
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.

Миопатия Дюшенна — наследственная патология, характеризующаяся прогрессирующим нарушением клеточного метаболизма в мышечной ткани, которое приводит к ее структурным изменениям и разрушению. Заболевание развивается только у мальчиков. Женщины – носители мутантного гена. Симптоматика болезни нарастает постепенно и начинает проявляться с самого детства. Родители замечают, что ребенок плохо стоит, с трудом карабкается на лесенку, часто падает во время ходьбы.
Миопатия была впервые описана в середине 19 века неврологом из Франции Г. Дюшенном. Он лечил больных с признаками мышечной патологии, анализировал наблюдения и представил свои умозаключения научному сообществу. Врач доказал генетическую природу заболевания.

Мышечная слабость прогрессирует постепенно. Симптомами заболевания являются: специфическая походка, неправильная осанка, позднее физическое и психическое развитие, утрата приобретенных двигательных навыков. У детей возникают проблемы с ходьбой и бегом. С возрастом признаки патологии становятся более выраженными и разнообразными. Симптомы усугубляются под воздействием негативных факторов – психофизического перенапряжения, острых инфекций, отравления. Они резко снижают качество жизни лиц с миопатией. У больных отмечается гипо- и арефлексия, искривление позвоночника, килевидная или седловидная грудная клетка, контрактуры суставов и ретракции сухожилий. Часто к поражению опорно-двигательного аппарата присоединяются проблемы с сердцем в виде кардиомиопатии и изменения интеллекта в виде олигофрении и дебильности.

Диагностика миодистрофии Дюшенна заключается в проведении медико-генетического исследования, которое позволяет выявить мутантный ген. Больным проводят биопсию мышечной ткани для определения в ней уровня белка дистрофина. Данное наследственное заболевание неизлечимо. Побороть недуг невозможно. Специалисты проводят симптоматическую и паллиативную терапию, облегчающую и продлевающую жизнь больным. Медикаментозное лечение глюкокортикостероидами, ЛФК, массаж и физиопроцедуры в комплексе дают относительно неплохие результаты.
Заболевание отличается тяжелым, злокачественным течением и неблагоприятным прогнозом. Больные рано перестают самостоятельно двигаться, с 8-10 лет используют костыли, а с 12 – инвалидные коляски. Шестнадцатилетние подростки испытывают нарушения со стороны органов дыхания и сердца, у некоторых снижается интеллект. Нарастающая мышечная слабость приводит к полной обездвиженности больного. Замедлить разрушение мышц невозможно, что связано с врожденным характером патологических изменений. Лица с миодистрофией умирают в возрасте 25-30 лет от инфекционной пневмонии или острой коронарной недостаточности.
Этиология

Миопатия Дюшена — генетически детерминированный недуг, наследуемый по рецессивному принципу, сцепленному с Х-хромосомой. Слабость мышц развивается у мальчиков. Девочки являются носителями мутантного гена. Именно они в 70% случаев передают патологию своим сыновьям. В оставшихся 30% мутации возникают спонтанно во время внутриутробного развития плода. Причины подобных мутаций неизвестны. Чаще всего это случается в близкородственных браках или у лиц, имеющих иные генетические аномалии.
Мутантный ген, обуславливающий развитие миопатии, получил название ген Дюшенна. Он отвечает за синтез белка дистрофина, который обеспечивает целостность мышечных волокон во время сокращения. Белок составляет основу миофибрилл, поддерживает их клеточный скелет, позволяет им активно и многократно сокращаться и расслабляться. У лиц с миопатией он отсутствует или вырабатывается в очень незначительном количестве. Мышечные волокна постепенно разрушаются и замещаются фиброзной тканью или жиром. В результате серьезно страдает функция движения у больных.
Симптоматика
Новорожденные дети не имеют каких-либо явных отклонений в здоровье и строении. Первые клинические признаки появляются у малышей 1,5-2 лет. Болезнь прогрессирует, а симптоматика нарастает.

проявления миопатии Дюшенна
- Родители замечают, что их ребенок двигается неловко и неустойчиво, постоянно спотыкается и падает при ходьбе, не может прыгнуть, подняться по ступенькам, встать из лежачего или сидячего положения. Больные дети очень медлительны и неуклюжи во время двигательной активности. Это связано с поражением мышц таза и ягодиц, которые становятся слабыми первыми.
- Появляется характерный симптом Говерса: больные, поднимаясь с пола, помогают себе руками – они опираются на свои колени и бедра.

Больные дети рано становятся инвалидами, прикованными к креслу. К 10-12 годам они полностью утрачивают способность к самостоятельному передвижению, а к 15 — к совершению любых действий. В течение пяти лет после этого атрофируются дыхательные мышцы.
Погибают больные обычно в возрасте 25 лет от неуклонно нарастающей слабости скелетных мышц, сформировавшейся стойкой дисфункции органов дыхания и сердечно-сосудистой системы.
Осложнения
Мышечная дистрофия Дюшенна сокращает жизненный путь человека. Это основное и самое страшное последствие болезни.
Осложнения со стороны опорно-двигательного аппарата:
- Остеопороз — уменьшение плотности костной ткани,
- Патология суставов — снижение их подвижности из-за сильной мышечной слабости,
- Сколиоз, кифоз, лордоз — различные формы искривления позвоночника.
Поражение органов пищеварения:
- Запоры — результат гиподинамии,
- Потеря веса из-за разрушения мышц,
- Нарушение процесса жевания и глотания требует питания больного через зонд.
- Поверхностное дыхание,
- Слабый кашлевой рефлекс,
- Частые ОРВИ,
- Недостаток кислорода в крови – головные боли по утрам, пробуждения по ночам, слабость, раздражительность, насыщенные сновидения.
У лиц с миопатией Дюшена развивается кардиомиопатия — слабость миокарда, проявляющаяся повышенной утомляемостью, одышкой, отеками ног, перебоями в работе сердца.
Своевременная диагностика и эффективная терапия могут отсрочить развитие осложнений или совсем предотвратить их появление.
Диагностические мероприятия
Диагностика синдрома Дюшенна не вызывает трудностей у специалистов, поскольку имеет весьма специфические симптомы.
Врачи традиционно начинают с опроса больного или его родителей и сбора анамнестических данных. Особое внимание они уделяют:
- Времени появления первых симптомов,
- Локализации первичной мышечной слабости,
- Общему самочувствию пациента,
- Наличию подобных расстройств у родных и близких.

Во время неврологического обследования выявляется:
- Слабость определенной группы мышц и определяется ее степень,
- Изменение мышечного тонуса,
- Атрофические процессы,
- Гипо- и арефлексия,
- Деформация стопы, груди, позвоночного столба.
Врачи наблюдают за больным ребенком, обращая внимание на то, как он ходит, бегает и встает с пола. Изменение походки — важный диагностический признак миопатии.
После проведения первичных диагностических процедур врачи могут заподозрить наличие у больного патологии и поставить предварительный диагноз. Чтобы его подтвердить или опровергнуть, пациента направляют на лабораторно-инструментальное обследование.
- Анализ крови на гормональный статус.
- Биохимический анализ крови на активность креатинкиназы – фермента, уровень которого у больных детей очень высок. Если КФК в норме, миопатию Дюшена исключают.
- Иммуногистохимическое исследование – микроскопия биоптата мышечной ткани, взятого от больного, с целью определения белка дистрофина. У лиц с миопатией он отсутствует.
- ДНК-тест – генетическое исследование крови больного, позволяющее определить мутантный ген и точно диагностировать патологию.
Дополнительные диагностические методики:
- Электрокардиография — выявление признаков поражения миокарда.
- Электромиография — определение фибрилляции, свидетельствующей о некрозе мышечных волокон. Эта методика оценивает состояние скелетной мускулатуры и подтверждает, что в основе патологии лежит именно поражение мышц, а не нарушение передачи нервных импульсов.
- Дыхательные пробы, рентгенография позвоночника и органов грудной клетки, УЗИ сердца — методы, не оказывающие существенного влияния на процесс постановки диагноза, но позволяющие выявить имеющиеся отклонения в структуре и функционировании органов дыхания и сердца.
Лечебный процесс
Болезнь Дюшенна, как и любая другая наследственная патология, неизлечима. Врачи проводят симптоматическую терапию, устраняющую неприятные проявления, предупреждающую развитие осложнений и продлевающую жизнь больных.

Комплексное поддерживающее лечение болезни:
Профилактика и прогноз
Супругам, в роду которых имелись случаи наследственных заболеваний, перед планированием беременности необходимо посетить врача-генетика. Профилактика патологии также заключается в проведении пренатальной диагностики. Выявив миопатию на ранних сроках, можно прервать беременность.

Миопатия Дюшена — наследственная патология, отличающаяся тяжелым течением и быстрым прогрессированием. Это современная медицинская проблема, характеризующаяся разрушением мышечной ткани и быстрым развитием мышечной слабости. Все без исключения больные погибают в раннем возрасте из-за развития не совместимых с жизнью осложнений. Только адекватная и комплексная терапия, четкое соблюдение рекомендаций врача, тщательный уход и забота родителей могут замедлить ход болезни.
Больные быстро становятся инвалидами и погибают в совсем юные годы. Самое страшное, что детям с миопатией не в силах помочь даже квалифицированные врачи, современные медицинские технологии и терапевтические методики 21 века. Болезнь до сих пор остается неизлечимой, забирая молодые жизни. Современные ученые-медики всего мира трудятся над созданием радикального способа борьбы с миопатией Дюшенна.
Видео: ребенок с синдромом Дюшенна
Читайте также: