
Мультифокальная моторная невропатия инвалидность
4 минуты Татьяна Григорьева 1025

- Причины инвалидности при полинейропатии
- Показания для прохождения МСЭ
- Критерии инвалидности
- Перечень необходимых обследований для МСЭ
- Видео по теме
Причин, способных привести человека к инвалидности, к сожалению, великое множество. Для удобства проведения МСЭ (медико-социальной экспертизы) их классифицируют, объединяя похожие нарушения интеллектуального, психического и физического здоровья в отдельно взятые категории, состоящие из более мелких подгрупп. К одной из них относятся полинейропатии или как их еще называют полирадикулоневропатии.
Это многочисленная группа, неоднородных по своей этиологии и некоторой симптоматике заболеваний, возникших вследствие воздействия негативных экзогенных либо эндогенных факторов.
Патологии объединяет обширное, как правило, дистальное симметричное поражение периферических нервов, приводящее к чувствительным, двигательным, вегетативно-сосудистым и трофическим нарушениям. Все это зачастую существенно снижает качество жизни пациента, тем самым приводя его к утрате трудоспособности в той или иной мере.
Причины инвалидности при полинейропатии
Заболевания данной группы, как уже упоминалось, могут развиваться на фоне воздействия различного ряда факторов, поэтому причинами получения инвалидности принято считать следующие:
- Общее заболевание.
- Инвалидность детства.
- Нетрудоспособность, наступившая из-за болезни, полученной во время прохождения военной службы, либо возникшая на протяжении 3-х месяцев после увольнения из ВС.
- Профессиональное заболевание: полинейропатия возникла под влиянием токсичных веществ в условиях производственной деятельности, под воздействием физических факторов (холодовая полинейропатия, вибрационная болезнь, вегетативная полинейропатия перенапряжения рук).
Показания для прохождения МСЭ
Лечащий врач направляет пациента для проведения МСЭ, если наблюдаются следующие отклонения здоровья, способные, по его мнению, стать препятствием для выполнения возложенных трудовых обязанностей. К ним относятся:
- Парезы и параличи рук и ног, устойчивый болевой синдром, выраженные трофические и вегетативные нарушения, сенситивная атаксия, признаки прогрессирующей вегетативной недостаточности, значительно ограничивающие жизнедеятельность человека.
- Затяжная временная нетрудоспособность при неблагоприятном либо сомнительном прогнозе, касающемся восстановления чувствительных, двигательных и других функций.
- Периодические рецидивы и прогрессирующее течение болезни с учетом ее происхождения, развитие постгерпетической невралгии.
- Отсутствие возможности возвращения к трудовой деятельности по специальности, обусловленное двигательным дефектом и/или условиями труда, противопоказанными для конкретного человека, если они не могут быть изменены по заключению комиссии.
Критерии инвалидности
После проведения медицинской экспертизы, определяющей состояние здоровья пациента и прогноз развития заболевания, ему может быть присвоена одна из трех групп инвалидности. Критерии, на которые опираются члены комиссии, при установлении степени нетрудоспособности следующие.

Что такое "Мультифокальная моторная нейропатия"?
- "Мультифокальная" - означает множественное поражение.
- "Моторная" - что в патологический процесс вовлекаются исключительно двигательные волокна нервов, ответственные за мышечную силу, выполнение движений.
- "Нейропатия" – это поражение периферических нервов.
Как часто встречается это заболевание среди населения?
Это очень редкое заболевание. Частота встречаемости ММН в популяции составляет всего 1–2 случая на 100 000 населения; диагностируется в 3-4 раза чаще у мужчин, а дебют (начало) заболевания приходится на трудоспособный возраст (20-50 лет).

Какой механизм развития данного заболевания?
ММН - хроническая множественная моторная нейропатия, в основе развития которой лежит аутоиммунный процесс, когда компоненты иммунной системы по ошибке начинают работать против структур собственного организма. В основе ММН лежит избирательное дизиммунное поражение нодальных зон моторных нервов - областей толстых нервных волокон, богатых ионными каналами и ответственных за передачу нервного импульса. В результате такого поражения развивается блок проведения возбуждения по двигательному нерву и движения нарушаются.
Что беспокоит пациентов при данном заболевании?
- асимметричная слабость мышц рук или рук > ног
- болевого синдрома и чувствительных нарушений нет!
ММН начинается у большинства пациентов с постепенного развития мышечной слабости, наиболее часто в руках. Нарушение чувствительности, болевой синдром отсутствуют.
Первоначальной жалобой может быть слабость в одной руке или в руках, значительно реже заболевание начинается с ног. Слабость в дистальных отделах рук (кистях) встречается в большинстве случаев и выявляется примерно у 95 % пациентов с MMН.
Более чем у половины больных отмечаются мышечные спазмы и подергивания. Поражение двигательных черепных нервов, ответственных за движение глаз, мышц лица, жевание, глотание и речь не отмечается. Тазовые функции не нарушаются.
Течение заболевания носит медленно прогрессирующий, либо волнообразный характер. Похудание мышц (гипо- или атрофия) нехарактерны и присутствуют на поздних стадиях заболевания, часто у пациентов, не получавших лечение. Признаков поражения центральной нервной системы нет.
На основании чего ставится диагноз и какие обследования могут быть мне назначены для его подтверждения?
В настоящий момент разработаны четкие критерии диагноза "Мультифокальная моторная нейропатия" (EFNS/PNS 2011). Согласно этим критериям достаточно анализа истории развития заболевания, неврологического осмотра и проведения электронейромиографии:
- электронейромиография позволяет обнаружить блоки проведения по двигательным волокнам периферических нервов в местах, нетипичных для их компрессии (вне туннелей), при этом параметры исследования сенсорных волокон неизменены. Кроме того, данное исследование исключает поражение других структур периферического нейро-моторного аппарата (двигательных нейронов, мышц).
- дополнительно может потребоваться нейровизуализация - УЗИ периферических нервов и МРТ плечевых сплетений с контрастированием, которые также высокоинформативны и помогают подтвердить диагноз и исключить иную патологию.
Однако на практике могут возникать трудности в постановке диагноза в случае, когда пациенты приходят на консультацию спустя годы от начала заболевания с грубыми двигательными нарушениями, атрофиями мышц или при атипичной форме болезни. В этих ситуациях требуется проведение дифференциального диагноза.

При проведении дифференциального диагноза список обследований расширяется:
- рутинные лабораторные исследования крови и мочи, МРТ позвоночника и спинного мозга не помогают в постановке диагноза "ММН", однако исключат возможные другие причины множественного поражения нервов (такие как сахарный диабет, ревматоидный артрит, вертеброгенная миелопатия, болезнь Хирояма и др.)
- анализ крови на антитела к ганглиозидам периферических нервов - антитела к GM1-ганглиозидам присутствуют примерно в половине случаев ММН
- генетический анализ крови на мутации гена PMP22 - исключит возможные наследственные причины нейропатии
- и т.д.
Конечно, не всегда назначаются все эти обследования. В большинстве случаев достаточно проведения только ЭНМГ-исследования. Следует подчеркнуть, что ЭНМГ-исследование должно быть проведено хорошо подготовленным и опытным специалистом на миорафе высокого класса. Методологические ошибки и недостаточный объем данного исследования часто приводят к ошибочным диагнозам. Поэтому мы рекомендуем проведение ЭНМГ в нашем центре.
Какое существует лечение?
ММН - хроническое заболевание, полное излечение невозможно. Но в настоящее время разработано эффективное лечение, способное контролировать заболевание. Терапия внутривенным иммуноглобулином (ВВИГ) является единственным методом лечения ММН, эффективность которого доказана в контролируемых исследованиях.
Эффективной дозой является 2 г препарата ВВИГ на 1 кг веса пациента (введение дозы за 3-5 дней). Подавляющее большинство пациентов положительно отвечают на данную терапию. Однако улучшение является кратковременным и требуется длительная поддерживающая терапия: периодические инфузии препаратов ВВИГ - по 1 г препарата ВВИГ на 1 кг веса пациента раз в 3-4 недели (введение за 3 дня) или введение 2 г препарата на кг веса раз в 1-2 месяца (введение за 3-5 дней).
Следует помнить, что не все препараты ВВИГ подходят для лечения ММН.
Основными требования к препаратам иммуноглобулина человеческого являются:
- безопасность препарата в отношении передачи инфекции;
- ВВИГ должен обладать хорошей переносимостью;
- содержание IgG должно быть не менее 95% общего содержания IgG;
- содержание IgA должно минимальным;
- препарат не должен проявлять тромбогенную (прокоагулянтную) активность;
- возможность проведения высокодозной терапии (нет ограничения суточной дозы);
- предпочтение отдается 10% растворам.
Противопоказаниями к терапии ВВИГ являются гиперчувствительность (аллергия) к компонентам ВВИГ, а также дефицит IgA. Поэтому перед проведением лечения рекомендуется исследование уровня IgA. При выявленном его дефиците лечение проводиться с осторожность, в частности используется премедикация и специальная подготовка.
При отсутствии терапии, нерегулярном введении ВВИГ или при лечении недостаточными дозами препарата двигательный дефицит может медленно, неуклонно и необратимо прогрессировать из-за вторичного повреждения аксонов.
Глюкокортикостероиды и плазмаферез неэффективны у пациентов с ММН!
Какой прогноз данного заболевания?
Приблизительно 80% пациентов отвечают на лечение ВВИГ. ММН не приводит к летальному исходу, так как в патологический процесс не вовлекаются мышцы участвующие в актах глотания и дыхания. Поскольку при ММН преимущественно поражаются мышцы рук, письмо и мелкая моторика могут быть ограничены. В этой связи следует рассмотреть альтернативные варианты профессиональной занятости.
Под наблюдением специалистов цПНС находится 40 пациентов с ММН.
Мы умеем диагностировать данное заболевание и определять оптимальный персонифицированный режим лечения препаратами ВВИГ.
Сотрудники центра заболеваний периферической нервной системы консультируют пациентов амбулаторно в рамках ОМС и на коммерческой основе.
Запись на приём: +7 (495) 374-77-76, +7 (985) 931-60-24
Множественная или мультифокальная двигательная или моторная невропатия – это относительно редкое заболевание нервной системы, которое имеет предположительно аутоиммунный тип и характеризующееся демиелинизацией двигательных волокон. Заболевание опасно тем, что ведет к инвалидизации без активного адекватного лечения.
Лечение мультифокальной двигательной нейропатии в клиниках Германии, Израиля, Австрии, США, Финляндии и Швейцарии проводится при помощи комплекса медикаментозных средств включающих, в том числе, самые современные иммуномодуляторы.
Диагностика мультифокальной моторной нейропатии за границей
Пациенты рекомендуемых нами неврологических клиник для подтверждения и уточнения диагноза проходят все необходимые диагностические тесты, включая:
- электронейромиографию, обеспечивающую выявление множественных парциальных блоков проведения по двигательным волокна при наличии нормальных проведений по все еще чувствительным волокнам;
- биопсии выявляющей субклинические изменения в волокнах;
- лабораторных анализов крови;
- игольчатой электромиографии выявляющей признаки денервации.
Моторная нейропатия
Поражение нервных стволов может приводить к нарушениям двигательной функции, утрате чувствительности. Диагностировать степень повреждения и устанавливать тип нейропатии трудно, поскольку клинические проявления, такие как слабость, атрофия мышц отсутствие рефлексов присущи многим заболеваниям затрагивающим, например мышцы или сухожилия.
К моторной нейропатии относят синдром Гийена-Баре, невропатию вызванную дифтерией, демиелинизирующую невропатию, идиопатическую плечевую плексопатию, порфирийную и мультифокальную нейропатию.
Мультифокальная моторная невропатия начинается с проблем, обнаруженных в ногах. Симптомы болезни проявляются в средней части голени. Но часто заболевание может затрагивать и верхние конечности, в области кистей одной или двух рук. Синдром Гийена-Барре и дифтерийная нейропатия – следствие острой инфекции, под влиянием которой поражаются не только нервы, но и корешки.
У больных отмечается слабость, повышается температура тела, мучают боли в руках и ногах, появляются явления парастезии, онемение и покалывание. Демиелинизирующая нейропатия по проявлениям схожа с синдромом Гийена-Барре. Это хроническое воспалительное заболевание, развивающееся на протяжении длительного периода которое не требует интенсивного лечения, иногда болезнь может отступить без вмешательства медиков.
Идиопатическая плечевая плексопатия возникает после травмы при падении на вытянутую руку. Провоцируют развитие патологии вывих плеча, ключичный перелом, перелом ребра и ещё многие повреждающие факторы. Редко возникает наследственная форма, сопровождается болью в надплечье и плече. При адекватном лечении человек выздоравливает через 2–3 года.
Чаще всего при нейропатии больные жалуются на слабость мышц, ограничения движений рук и ног, боли по ходу нерва, онемение, жжение, мурашки, нарушение чувствительности на участке пораженного нерва, причем ощущения могут быть снижены или повышены. Обычно специалисты проводят исследования по результатам анализов, учитывая первоначальные симптоматические проявлений болезни.
Окончательный диагноз ставится на основании всех тщательно изученных медицинских обследований, выяснения причин болезни и степень её тяжести. Неврологическое обследование включает тестирование рефлексов и выявление активности чувствительных и двигательных функций нервов.
Мультифокальная моторнавя нейропатия вовлекает в патологический процесс сенсорные волокна. В основе болезни лежит аутоиммунный фактор, способствующий демиелинизации периферических нервов. Дифференциальная диагностика часто затрудняется из-за мышечных подергиваний (фасцикуляций), судорог в икроножных мышцах (крампи).
В более поздних стадиях наблюдается атрофия мышц. В случае несвоевременной медицинской помощи больной может стать инвалидом. Для исследований данного заболевания используют клинические, лабораторные результаты. Применяют электромиографические и иммунологические данные.
Кроме лечения определенными лекарствами применяют физиотерапию, массаж, отличные результаты показывает лечебная гимнастика. В процессе лечения о его эффективности можно судить по положительной динамике увеличения силы мышц нижних и верхних конечностей. Из анализа крови невозможно узнать о наличии нейропатии.
Но такое исследование необходимо для того, чтобы можно было определить причины, выявить сопутствующие болезни вызвавшие нейропатию, например сахарный диабет или дефицит витаминов группы В. Методы диагностики такие как рентгенография, компьютерная томография, магнитно-резонансное исследование позволяют выявить патологические изменения в нервных волокнах.
Синдром нейропатии — это совокупность симптомов с общим патогенезом. При выявлении вида нейропатии необходимо учитывать все результаты исследований, чтобы определить эффективные методики лечения заболевания. Только специалист может разобраться в этих вопросах, как правило, заболевание невозможно устранить самостоятельно. Для восстановления работоспособности пораженных частей тела понадобится специальный курс терапии. Лечение может быть быстрым или длительным, все зависит от степени поражения нерва, больным рекомендуется набраться терпения.
Эксперт-редактор: Мочалов Павел Александрович | д. м. н. терапевт
Организация лечения мультифокальной моторной нейропатии за границей
является лидером в оказании услуг по медицинскому менеджменту. Одной из наших сильных сторон является тесное сотрудничество с известными медицинскими центрами, занимающимися, в том числе, лечением неврологических заболеваний.
Мы организовываем лечение у опытных докторов, которые могут предложить своим иностранным пациентам высококвалифицированные услуги при адекватной стоимости лечения и великолепном сервисе. Очень важным плюсом сотрудничества с нашей компанией, является возможность сократить ожидание приема у необходимого специалиста до минимума.
Диагностика и лечение сенсомоторной нейропатии
Такое обследование может провести и эндокринолог, и невролог.
Какие нарушения чувствительности выявляются и как?
— Нарушения тактильной чувствительности, то есть чувства прикосновения, давления.
Используется монофиламент – прибор, напоминающий карандаш, на конце которого есть тонкая нить весом 10г, им прикасаются к различным участкам тела (например, к стопам)
— Нарушения болевой чувствительности.
Выявляются с помощью несильного укола тыльной поверхности большого пальца специальной иглой с притупленным концом
— Нарушения температурной чувствительности.
Их можно обнаружить при помощи определения разницы в ощущениях тепла и холода специальным инструментом
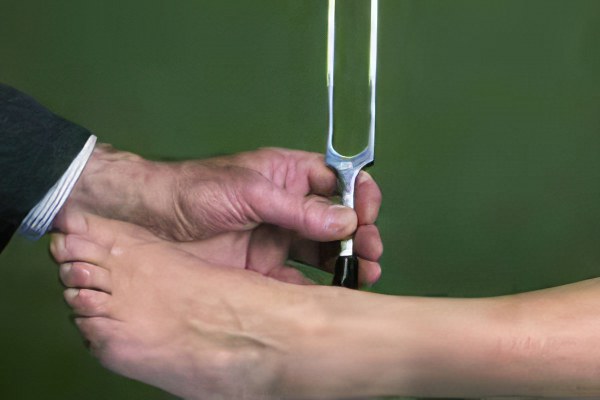
— Нарушения вибрационной чувствительности.
Определяются с использованием камертона
Определяются с помощью неврологического молоточка, оцениваются рефлексы – коленные и ахилловы.
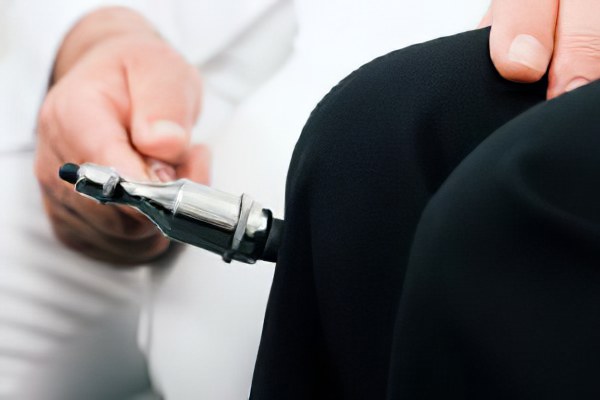
Более точную информацию дает электронейромиография (ЭНМГ) — это комплексное исследование, которое позволяет выявить изменения в нервной ткани на самых ранних стадиях. Изучается скорость проведения нервного импульса по нерву и то, как на этот импульс реагирует мышца. Процедура электронейромиографии проводится в положении лежа или сидя. На кожу устанавливается электрод аппарата, затем в мышцу погружается игла электрода. Электрод при этом подключен к прибору, улавливающему сигналы от мышцы к электромиографу. Запись мышечной активности напоминает электрокардиограмму. Исследование длится от 15 до 90 минут, в зависимости от количества обследуемых мышц. Оно не оказывает негативного влияния на сам нерв, но может вызывать неприятные ощущения во время проведения процедуры.
Биопсия нерва – иссечение небольшой его части с последующим изучением структуры под микроскопом — является наиболее точным методом диагностики полинейропатии. Однако в повседневной практике этот метод применяется нечасто.
Боли в ногах, онемение и даже судороги могут возникать и при проблемах с сосудами. Для того, чтобы разрешить, в чем причина – в сосудах или нервной системе — проводится ультразвуковое дуплексное сканирование (УЗДГ) артерий и/или голеней и стоп.
Основа лечения – длительная, стабильная компенсация диабета Достижение целевого гликированного гемоглобина позволяет снизить риск на 50%! (по данным исследования DCCT). Нормализация глюкозы крови часто приводит к устранению нейропатических болей, в то время как лекарственная терапия на фоне декомпенсации углеводного обмена может быть мало – или вовсе неэффективной.
Есть одно важное правило- снижение уровня гликированного гемоглобина не должно быть слишком быстрым и резким– это может только ухудшить ситуацию и вызвать/усилить боль.
Лекарственные препараты
— Препараты альфа-липоевой кислоты (октолипен, берлитион, тиогамма, тиоктацид) широко применяются в России. Считается, что они действуют как антиоксиданты, снижая выработку тех самых свободных радикалов, которые образуются из-за избытка глюкозы. Но как вы понимаете, если будет сохраняться высокий уровень глюкозы крови, они не будут эффективны. — Препараты витаминов группы В (тиамина, бенфотиамина и других) – применяются для восстановления нервного волокна и его оболочки. Но опять же, необходимы соответствующие условия – компенсация сахарного диабета. — При сильной боли назначаются препараты для ее снижения или устранения. Они не лечат, лишь снимают симптом, но иногда они просто необходимы, ведь боль может быть очень сильной и не давать человеку спокойно жить. Что к ним относится?
— некоторые виды антидепрессантов, — противосудорожные препараты (габапентин, прегабалин, карбамазепин), — в случае неэффективности терапии болевого синдрома вышеуказанными лекарственными средствами, возможно назначение опиоидных анальгетиков.
Данные препараты назначаются строго врачом и выписываются по рецепту. — При нарушении липидного обмена может нарушаться и кровоснабжение нервов, что также ухудшает течение нейропатии. Для нормализации уровня холестерина врач назначает специальные препараты (статины, фенофибрат).
Диагностика
Как и при многих других болезнях, диагностика нейропатии начинается со сбора анамнеза. Врач должен выяснить причину, которая стала пусковым механизмом для данной болезни. Это может осложняться тем, что заболевание или травмы были за много месяцев до появления первых симптомов. Поэтому стоит подробно расспрашивать про перенесенные заболевания (особенно вирусной этиологии), назначении новых лекарственных препаратов, вредных условиях труда, злоупотреблении алкоголем. Также важно выяснить наличие похожих симптомов у родственников.
Характер первых симптомов также немаловажен. Поэтому пациент должен детально рассказать про дебют заболевания: когда началось, в какой части тела, первые проявления, их длительность и прогрессирование.
Осмотр невропатолога включает в себя проверку всех видов чувствительности, наличие рефлексов, степени поражения мышц.
К стандартам также относятся анализы биологических жидкостей, рентген грудной клетки, измерение уровня глюкозы. Дополнительные методы исследования применяют на основании уже полученных данных.
Поражение нервных стволов может приводить к нарушениям двигательной функции, утрате чувствительности. Диагностировать степень повреждения и устанавливать тип нейропатии трудно, поскольку клинические проявления, такие как слабость, атрофия мышц отсутствие рефлексов присущи многим заболеваниям затрагивающим, например мышцы или сухожилия.
К моторной нейропатии относят синдром Гийена-Баре, невропатию вызванную дифтерией, демиелинизирующую невропатию, идиопатическую плечевую плексопатию, порфирийную и мультифокальную нейропатию.
Мультифокальная моторная невропатия начинается с проблем, обнаруженных в ногах. Симптомы болезни проявляются в средней части голени. Но часто заболевание может затрагивать и верхние конечности, в области кистей одной или двух рук. Синдром Гийена-Барре и дифтерийная нейропатия – следствие острой инфекции, под влиянием которой поражаются не только нервы, но и корешки.
У больных отмечается слабость, повышается температура тела, мучают боли в руках и ногах, появляются явления парастезии, онемение и покалывание. Демиелинизирующая нейропатия по проявлениям схожа с синдромом Гийена-Барре. Это хроническое воспалительное заболевание, развивающееся на протяжении длительного периода которое не требует интенсивного лечения, иногда болезнь может отступить без вмешательства медиков.
Идиопатическая плечевая плексопатия возникает после травмы при падении на вытянутую руку. Провоцируют развитие патологии вывих плеча, ключичный перелом, перелом ребра и ещё многие повреждающие факторы. Редко возникает наследственная форма, сопровождается болью в надплечье и плече. При адекватном лечении человек выздоравливает через 2–3 года.
Чаще всего при нейропатии больные жалуются на слабость мышц, ограничения движений рук и ног, боли по ходу нерва, онемение, жжение, мурашки, нарушение чувствительности на участке пораженного нерва, причем ощущения могут быть снижены или повышены. Обычно специалисты проводят исследования по результатам анализов, учитывая первоначальные симптоматические проявлений болезни.
Окончательный диагноз ставится на основании всех тщательно изученных медицинских обследований, выяснения причин болезни и степень её тяжести. Неврологическое обследование включает тестирование рефлексов и выявление активности чувствительных и двигательных функций нервов.
Мультифокальная моторнавя нейропатия вовлекает в патологический процесс сенсорные волокна. В основе болезни лежит аутоиммунный фактор, способствующий демиелинизации периферических нервов. Дифференциальная диагностика часто затрудняется из-за мышечных подергиваний (фасцикуляций), судорог в икроножных мышцах (крампи).
В более поздних стадиях наблюдается атрофия мышц. В случае несвоевременной медицинской помощи больной может стать инвалидом. Для исследований данного заболевания используют клинические, лабораторные результаты. Применяют электромиографические и иммунологические данные.
Кроме лечения определенными лекарствами применяют физиотерапию, массаж, отличные результаты показывает лечебная гимнастика. В процессе лечения о его эффективности можно судить по положительной динамике увеличения силы мышц нижних и верхних конечностей. Из анализа крови невозможно узнать о наличии нейропатии.
Но такое исследование необходимо для того, чтобы можно было определить причины, выявить сопутствующие болезни вызвавшие нейропатию, например сахарный диабет или дефицит витаминов группы В. Методы диагностики такие как рентгенография, компьютерная томография, магнитно-резонансное исследование позволяют выявить патологические изменения в нервных волокнах.
Синдром нейропатии - это совокупность симптомов с общим патогенезом. При выявлении вида нейропатии необходимо учитывать все результаты исследований, чтобы определить эффективные методики лечения заболевания. Только специалист может разобраться в этих вопросах, как правило, заболевание невозможно устранить самостоятельно. Для восстановления работоспособности пораженных частей тела понадобится специальный курс терапии. Лечение может быть быстрым или длительным, все зависит от степени поражения нерва, больным рекомендуется набраться терпения.

Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность - "Лечебное дело" в 1991 году, в 1993 году "Профессиональные болезни", в 1996 году "Терапия".
Наши авторы

Медико-социальная экспертиза и инвалидность при нервно-мышечных заболеваниях
Нервно-мышечные заболевания — основная группа наследственной патологии нервной системы. Если учитывать широкий спектр патологии мышечной системы, то их распространенность в популяции в зависимости от формы составляет 1—6 на 100 000 населения.
За последние годы благодаря применению новых современных методов исследования во многом продвинулось изучение этих заболеваний. Так, электронная микроскопия позволила выделить целую группу врожденных непрогрессирующих миопатий (нитевидная, митохондриальная и др.), ЭМГ, ЭНМГ внесли много нового в диагностику спинальных и невральных амиотрофий. Развитие методов молекулярной генетики позволило определить локализацию мутантных генов, осуществить их клонирование при основных наследственных нервно-мышечных заболеваниях. Это сделало возможным выявление носительства заболевания среди здоровых родственников больных, осуществление пренатальной диагностики. Достижения нейрогенетики наметили реальные пути профилактики и лечения этих тяжелых заболеваний. Серьезное внимание обращается на фенокопии мышечных заболеваний (симптоматические экзогенные миапатии, миотонии, миастении) при эндокринных, аутоиммунных, соматических болезнях, интоксикациях, пирамидных, экстрапирамидных видах патологии нервной системы, что имеет важное клиническое, в частности диагностическое и прогностическое значение.
Заболевания с преимущественным поражением нервно-мышечного
аппарата различаются типом наследования, морфологическими, биохимическими характеристиками, возрастом дебюта, преимущественным поражением тех или иных групп скелетных мышц, характером течения, темпом прогрессирования.
Условно их можно разделить на 3 группы:
1.Первичные (миопатии), когда процесс начинается с мышцы. Среди них выделяют прогрессирующие мышечные дистрофии, врожденные непрогрессирующие метаболические и некоторые другие миопатии.
2.Вторичные, когда первично поражаются нервы и мотонейроны передних рогов спинного мозга. Они разделяются на спинальные (первично страдают нейроны) и невральные (патологический процесс преимущественно локализуется в проводниках периферического двигательного нейрона). Атрофия мышц в этой группе — явление вторичное.
3.Заболевания, при которых первичным предполагается поражение нервно-мышечного синапса (миастения, миотония, миоплегия).
Подобная рубрификация помогает ориентироваться в обширной группе нервно-мышечных заболеваний, важна с позиций практической диагностики. Вместе с тем гистологические исследования показывают, что первичное или вторичное поражение мышц не исключает распространения процесса на все звенья нервно-мышечного аппарата (Дунаевская Г. Н., 1997).
Вторичные наследственные невропатии, проявляющиеся мышечными атрофиями, в настоящее время чаще рассматриваются среди заболеваний периферической нервной системы (в группе наследственных моторных и сенсорных невропатий).
Общая классификация нервно-мышечных заболеваний весьма сложна.
Существуют рубрификации отдельных видов нервно-мышечной патологии, среди которых, в свою очередь, основное внимание обращается на наиболее часто встречающиеся клинические формы. Ниже приводится несколько адаптированная классификация болезней скелетных мышц (миопатий), разработанная Walton (1990).
А. Генетически обусловленные миопатии:
I. Мышечные дистрофии:
а) псевдогипертрофические типы:
— рецессивный, сцепленный с Х-хромосомой (тяжелый — тип Дюшенна);
— рецессивный, сцепленный с Х-хромосомой (легкий — тип Беккера);
б) лицелопаточно-плечевые типы (аутосомно-доминант- ные, Ландузи-Дежерина и др.);
в) конечностно-поясной тип (аутосомно-рецессивный, Эрба-Рота);
г) дистальные типы (аутосомно-доминантные):
— поздний (Веландер);
— юношеский (Бимонд, Барнес);
д) глазной тип (аутосомно-доминантный);
е) глазо-глоточный тип.
II. Конгенитальные миопатии:
а) болезнь центрального стержня;
б) немалиновая (нитевидная);
в) миотубулярная или центронуклеарная;
г) митохондриальная;
д) доброкачественная гипотония.
III. Миотонии:
1) миотоническая дистрофия (аутосомно-доминантная);
2) конгенитальная миотония (аутосомно-доминантная, болезнь Томсена);
3) конгенитальная парамиотония (аутосомно-доминантная, болезнь Эйленбурга) и др.
IV. Гликогенозы:
дефицит мышечной фосфорилазы (аутосомно-доминантная, болезнь Мак Ардля) и др.
V. Липоидозы:
дефицит карнитина и др.
VI. Периодические параличи:
1) гипокалиемический;
2) гиперкалиемический;
3) нормокалиемический.
Б. Экзогенные (ненаследственные) миопатии:
I. Эндокринные и метаболические:
1) гипертиреоидная;
2) гипотиреоидная;
3) болезнь Иценко-Кушинга с миопатией;
4) акромегалия с миопатией;
5) болезнь Аддисона с миопатией;
6) гиперпаратиреоидизм с миопатией;
7) алкогольная;
8) стероидная и др.
II. Карциноматозная миопатия.
III. Тяжелая миастения с миопатией.
IV. Полимиозиты:
1) первичный (идиопатический) полимиозит (органоспецифическое аутоиммунное заболевание);
2) вторичные полимиозиты или дерматомиозиты при болезнях
соединительной ткани;
3) полимиозиты при паразитарных заболеваниях.
V. Опухоли мышц (рабдомиомы, рабдосаркомы).
Двигательная функция имеет универсальное значение в жизни человека. Ее нарушение приводит к социальной недостаточности, ограничению жизнедеятельности, в частности способности к труду. Большинство этих болезней относится к наследственным, что обусловливает неуклонное прогрессирование, трудности лечения и реабилитации. Тяжелая инвалидность наблюдается почти при всех нервно-мышечных заболеваниях. Например, при миодистрофиях инвалидами II или I группы признаются около 60 % больных, при спинальных амиотрофиях — 40—50%. Среди больных миастенией лишь 20 % полностью нетрудоспособны. Социальное значение усугубляется молодым, работоспособным возрастом больных, нарастанием тяжести инвалидности в связи с прогрессированием заболевания.
Определение
Прогрессирующие мышечные дистрофии (ПМД) — группа наследственных заболеваний с первичным поражением мышц вследствие генного дефекта, характеризующихся неуклонно прогрессирующим течением.
1.Основные подходы к неврологической диагностике клинических форм ПМД:
— семейный анамнез, тип наследования (изучение родословных);
— возраст манифестации болезни;
— характер и темп прогрессирования;
— своеобразие топографии мышечных атрофий и последовательность их распространения.
Дифференциальная диагностика
Проводится главным образом с другими нервно-мышечными заболеваниями:
1.С врожденными (непрогрессирующими) миопатиями. Проявляются с рождения или в раннем детском возрасте. Их формы определяются патоморфологическими изменениями мышц, выявляемыми при электронной микроскопии (болезнь центрального стержня, немалиновая, центрально-ядерная и другие формы мио- патий). Наследование чаще по аутосомно-доминантному типу. Диагностическое значение имеют: выраженная генерализованная гипотония после рождения, преимущественное поражение мышц тазового пояса и ног, задержка моторного развития, сохранность интеллекта, частое сочетание с врожденным вывихом бедра, кифосколиозом, конской стопой; активность КФК нормальна. Главная особенность — отсутствие прогрессирования, очень медленное прогрессирование, иногда даже улучшение двигательных функций с возрастом. Ограничение жизнедеятельности вследствие нарушения способности к передвижению может быть незначительным или умеренным. При правильной профориентации и рациональном трудоустройстве больные нередко могут доработать до пенсии по возрасту. Однако на протяжении всей жизни они имеют ряд ограничений в учебе, выборе профессии, трудовой и социальной адаптации, что определяет необходимость социальной защиты, а иногда определения инвалидности.
2.С митохондральными миопатиями (энцефаломиопатиями). Они выделяются из группы непрогрессирующих миопатий, так как митохондриальный дефект (нарушение окислительного фос- форилирования) не ограничивается мышечной тканью, а вызывает и поражение ЦНС. Болезнь начинается в любом возрасте, но чаще у детей и подростков, с поражения экстраокулярных мышц (птоз без диплопии) и может сопровождаться генерализованной мышечной слабостью, плохой переносимостью даже умеренных физических нагрузок. У многих больных наблюдаются церебральные симптомы: эпилептические припадки, инсультоподобные эпизоды, мигренеподобные приступы, миоклонии, мозжечковая атаксия, снижение интеллекта, атрофия зрительного нерва, тугоухость, а также поражение внутренних органов: кардиомиопатия, почечная тубулопатия, патология печени и эндокринных желез. Многообразие нарушений функций определяет характер и степень ограничения жизнедеятельности, частоту и тяжесть инвалидности. Верификация диагноза на основании биопсии мышц (характерные морфологические изменения), увеличения содержания лактата в плазме крови и ликворе (особенно после физической нагрузки).
3.С другими формами метаболических миопатий, например гликогенозными (болезнь Мак-Ардля), эндокринными, воспалительными (полимиозит, дерматомиозит).
Эндокринные миопатии наиболее характерны для патологии щитовидной железы. Диагностика определяется выявлением диффузного токсического зоба или гипотиреоза, подтверждается результатами гормональных исследований, типичными для миопатии данными ЭМГ, эффективностью тиреостатической терапии мерказолилом, а при гипотиреозе тиреоидином.
Диагностика полимиозита или дерматомиозита основывается на наличии в картине болезни общеинфекционных проявлений, похудения, изменений кожных покровов. Атрофии локализуются преимущественно в передней группе мышц шеи, плечевого и тазового пояса. Типичны ретракции, болевой синдром (наиболее отчетливый в мышцах голеней, четырехглавой мышце бедра, трапециевидной). Изменения ЭМГ специфичные для полимиозита: снижение амплитуды при высокой частоте разряда, медленные потенциалы. При исследовании мышечных биоптатов выявляются характерные воспалительные изменения, что позволяет верифицировать диагноз.
Саркоидозная миопатия, представляющая собой гранулематозный полимиозит, наблюдается у 10—20% больных саркоидозом. Клинически — гипотрофия и слабость мышц преимущественно проксимального отдела конечностей, туловища, нередки боли, особенно в мышцах спины. В мышцах могут пальпироваться множественные плотные бугорки (конгломераты саркоидных гранулем). Исследование биоптата мышц с выявлением полинуклеарных гигантоклеточных гранулем делает диагноз несомненным.
4.С вторичными неврогенными амиотрофиями (невральными, спинальными)
Течение и прогноз
Определяются генетическим фактором, а следовательно, клиническими особенностями. Применение ДНК-диагностики дает возможность прогноза течения и исходов заболевания в каждом конкретном случае. Сведения о примерных сроках начала, продолжительности заболевания при наиболее значимых клинических формах миодистрофии приведены выше. В целом формы ПМД с доминантным типом наследования протекают благоприятнее, позднее манифестируют, чем с рецессивным.
Целесообразно выделять стадии течения миодистрофического процесса (Бадалян Л. О., 1974):
I стадия — с умеренно выраженными двигательными нарушениями (больные могут ходить, выполнять легкую работу, слабость выявляется при нагрузке);
II стадия — с выраженными двигательными затруднениями при ходьбе, подъеме по лестнице, при выполнении физической работы;
III стадия — паралитическая: грубые контрактуры, деформации, самостоятельное передвижение невозможно.
Важное социальное значение имеют темп прогрессирования заболевания и возраст больного к его началу. Известно, что больные с ПМД Ландузи-Дежерина, Беккера, при позднем начале формы Эрба-Рота часто могут до начала болезни получить образование, специальность, что во многом определяет их реабилитационный потенциал, позволяет уменьшить степень социальной недостаточности.
Можно говорить о вариантах течения патологического процесса:
1) неблагоприятный вариант — обездвиженность через 5—10 лет от начала болезни, быстрое нарастание степени нарушения жизнедеятельности, тяжести инвалидности;
2) средний темп прогрессирования — через 10—15 лет имеются выраженные двигательные нарушения, прогноз в отношении возможности самообслуживания плохой;
3) медленный темп прогрессирования — через 10—15 лет от начала болезни нет выраженных двигательных нарушений, больной свободно передвигается, трудовой прогноз на ближайшие годы относительно благоприятен, возможна частичная трудовая адаптация, иногда оправдано профессиональное обучение.
Читайте также: