
Наследственная мозжечковая атаксия пьера мари лечение

- 29 Июня, 2018
- Неврология
- Евдокимова Ирина
Мозжечковая атаксия Пьера-Мари — это тяжелое наследственное заболевание. Но в отличие от многих других генетических патологий, недуг впервые проявляется не в детском возрасте, а у взрослых людей. С течением времени симптомы нарастают, а заболевание прогрессирует.
Пути наследования патологии
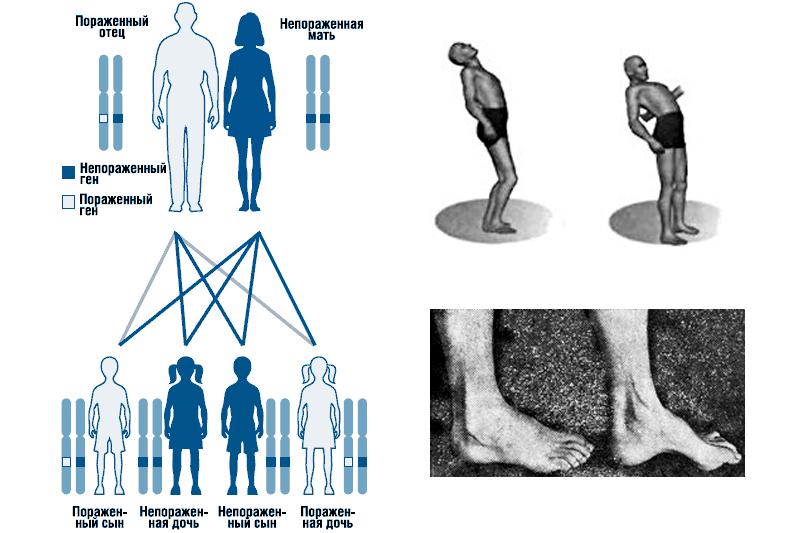
Наследственная мозжечковая атаксия Пьера-Мари передается детям от больного родителя. При этом вероятность рождения ребенка с патологией составляет 50%. Заболеванию одинаково подвержены и девочки, и мальчики.
Если в семье родился ребенок с таким заболеванием, то в будущем он может передать недуг своим детям. Опасность патологии заключается в том, что на момент деторождения человек может даже не подозревать о том, что он болен. Первые признаки мозжечковой атаксии Пьера-Мари проявляются в довольно зрелом возрасте. Здоровые дети не являются носителями дефектного гена и не передают заболевание потомству. Такой вид наследования генетики называют аутосомно-доминантным.

Причины заболевания
Непосредственными причинами мозжечковой атаксии Пьера-Мари являются патологические изменения в коре и ядрах мозжечка. Этот орган в организме человека отвечает за равновесие тела и обеспечивает координацию движений при перемещении.
У больного человека клетки мозжечка подвергаются дегенерации. В результате орган уменьшается в объеме. Такие же изменения претерпевают и другие отделы центральной нервной системы, например, варолиев мост, который отвечает за передачу спинальных импульсов. Дегенеративные процессы происходят в продолговатом мозге и в проводящих путях спинного мозга. Заболевание поражает практически все системы организма, отвечающие за равновесие и движение тела.
На поздних стадиях болезни поражаются глазные нервы, что приводит к ухудшению зрения.
Как проявляется болезнь?
Симптомы мозжечковой атаксии Пьера-Мари возникают обычно после 30-ти лет. Можно выделить следующие признаки патологии:
- Болезнь всегда начинается с нарушения походки. Человек вынужден широко расставлять ноги, чтобы поддерживать равновесие тела. Заметно сильное пошатывание при ходьбе, больному особенно трудно совершать повороты. Пациент идёт неуверенно, иногда падает. Это результат не только расстройства равновесия, но и плохой слаженности движений. В дальнейшем нарушения координации наблюдаются не только при ходьбе, но и в стоячем положении.
- Затем нарушаются движения рук. Больному становится трудно дотянуться до предмета. Пациент часто промахивается рукой мимо цели. Мелкая моторика затруднена. По этой причине ухудшается почерк, он становится размашистым и нечетким. При движениях наблюдается тремор верхних конечностей.
- Изменяется речь больного. Пациент разговаривает медленно, разделяя слова на слоги и выделяя их ударением или особой интонацией. Такую речь неврологи называют скандированной.
- Мимика становится бедной. Выражение лица — застывшее, маскообразное.
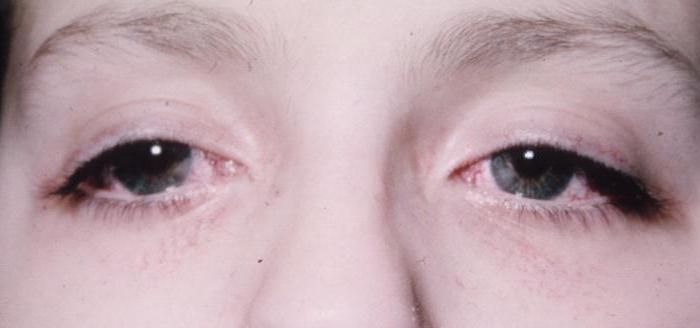
- Очень часто наблюдается дрожание глазных яблок — нистагм. Возникают и другие нарушения органа зрения — опущение верхнего века (птоз), паралич глазных мышц, косоглазие, расстройства бинокулярности. Из-за этого человек ощущает двоение в глазах. При поражении зрительных нервов отмечается сужение полей восприятия. Пациент способен видеть не все предметы, которые находятся перед ним. Довольно часто возникает катаракта.
- Мышечный тонус падает, конечности становятся слабыми. Со временем возникают боли в ногах и пояснице. Периодически происходят подёргивания мышц.
- При болезни Пьера-Мари нередко страдает психика человека. Сначала возникают трудности с запоминанием информации. Часто появляется депрессивное состояние. В дальнейшем это приводит к снижению интеллекта. Такие признаки наблюдаются не у всех больных. Но примерно в половине случаев происходит снижение умственных способностей.
Такая характеристика мозжечковой атаксии Пьера-Мари говорит о том, что это очень серьезное и тяжелое заболевание. Оно неуклонно прогрессирует. Течение патологии усугубляют инфекционные болезни, умственное или физическое напряжение.

Диагностика болезни
При появлении признаков атаксии необходимо обратиться к неврологу. При внешнем осмотре больного выявляется нистагм и другие глазодвигательные отклонения. Заметны речевые нарушения. Врач проводит пальце-носовую пробу: больному предлагают с закрытыми глазами дотронуться пальцем до кончика носа. Пациент с атаксией обычно не может попасть в цель. При постукивании неврологическим молоточком по сухожилию определяются повышенные рефлексы.
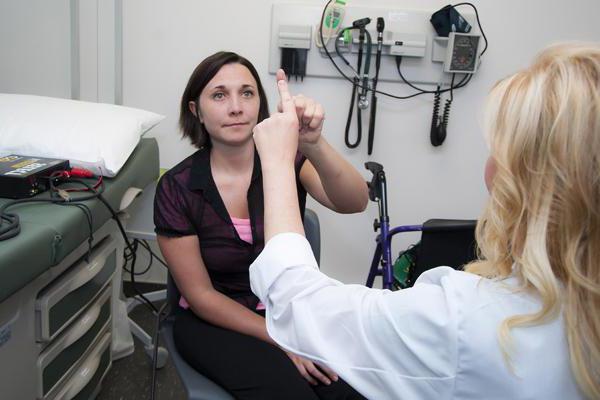
Врач может назначить дополнительные виды обследований, чтобы выявить происхождение атаксии:
- МРТ и КТ головного мозга;
- УЗИ головного мозга.
В результатах исследований видна атрофия мозжечка. Кроме этого, врач собирает анамнез. Необходимо уточнить, встречались ли у членов семьи подобные заболевания. Это поможет сделать вывод о наследственном характере атаксии.
Выявить нарушения координации движений несложно. Труднее отделить мозжечковую атаксию Пьера-Мари от других подобных болезней.
Дифференциальный диагноз
Существует множество мозжечковых атаксий и других болезней, сопровождающихся расстройством координации движений, речи и зрения. Врачу необходимо провести дифференциальную диагностику этих состояний.
Похожие проявления встречаются при другом наследственном заболевании - атаксии Фридрейха. Но эта патология проявляется в более молодом возрасте. При болезни Пьера-Мари повышены сухожильные рефлексы, а при атаксии Фридрейха - снижены. Отличается и тип наследования недуга. Болезнь Фридрейха может передаваться потомкам от вполне здоровых родителей, которые являются носителями дефектных генов.
Другим схожим заболеванием является рассеянный склероз. Но эта патология тоже проявляется в более молодом возрасте. Отличить эти две болезни поможет МРТ-обследование. При рассеянном склерозе будут видны очаги демиелинизации нейронов мозга. Этого не наблюдается при атаксии Пьера-Мари.
Излечимо ли заболевание?
На сегодняшний день это заболевание является неизлечимым. Невозможно подобрать такую терапию, которая остановила бы процесс дегенерации клеток мозжечка. Врач может предложить только симптоматическое лечение. Применяются следующие виды препаратов:
- Ноотропные лекарства ("Пирацетам", "Циннаризин", "Кавинтон"). Они улучшают память и уменьшают головокружение.
- Антидепрессанты назначают при расстройствах психики.
- Витамины группы В нормализуют работу мозга.
Эти медикаменты не способны полностью излечить мозжечковую атаксию Пьера-Мари. Они помогут лишь смягчить симптоматику и замедлить развитие болезни.
Лечебная гимнастика
Больным назначают лечебную гимнастику для укрепления мышц и улучшения скоординированности движений.
Пациентам с атаксией Пьера-Мари полезны упражнения на равновесие. Сначала задания выполняют в сидячем положении. Это может быть тренировка точности движений, повороты тела, подъем и перемещение предметов. Постепенно упражнения усложняют, уменьшая плоскость опоры сидения.

Затем человек совершает движения ногами, держась руками за брусья. Когда больному удается держать равновесие в такой позе, можно переходить к ходьбе. Пациент пытается ходить, совершая движения руками, двигаясь спиной или боком.
Больным с глазодвигательными нарушениями полезна гимнастика для органа зрения. Пациент пробует фиксировать свой взгляд на определенном предмете или точке, при этом меняя положение головы и тела.
Инвалидность при мозжечковой атаксии
При атаксии Пьера-Мари человек частично или полностью теряет трудоспособность. Поэтому он вправе получить группу инвалидности.
Больной не в состоянии выполнять многие виды работ. Из-за атаксии человек не может длительное время находиться на ногах. Нарушение координации рук препятствует работе, требующей точности движений. Проблемы с речью мешают деятельности, связанной с общением (например, с клиентами). Если болезнь осложнена интеллектуальными нарушениями, то человек не может заниматься умственным трудом. Симптоматика заболевания накладывает большие ограничения на работу пациента.
Степень инвалидности устанавливается индивидуально, в зависимости от состояния больного.

Прогноз заболевания
Атаксия Пьера-Мари - это прогрессирующее заболевание. С течением времени симптомы нарастают. Можно сказать, что у этого недуга неблагоприятный прогноз.
Болезнь не влияет на продолжительность жизни и не приводит к летальному исходу. Но изменения в мозжечке необратимы, и со временем пациент теряет трудоспособность.
На сегодняшний день медицина не может вылечить это заболевание, как и многие другие генетические патологии. Однако врачебная помощь поможет несколько замедлить развитие недуга.
Наследственная мозжечковая атаксия Пьера-Мари — генетико-семейное заболевание, обусловленное прогрессирующим мозжечковым расстройством, отягощенным поражением пирамидальных путей. Характеризуется повышенным рефлексом сухожилий, скандированной речью, дисбалансом координации движений, нарушением зрения и глазодвигательной моторики. Характер наследования — аутосомно-доминантный. Мутантный ген имеет высокую популяцию: пропуск поколений встречается редко.
- Причины и течение атаксии Пьера-Мари
- Симптомы атаксии Пьера-Мари
- Дифференциальная диагностика атаксии Пьера-Мари
- Диагностика атаксии Пьера-Мари
- Лечение и прогноз атаксии Пьера-Мари
Среди наследственных болезней спиноцеребеллярные атаксии стоят по частоте заболеваемости на втором месте после нервно-мышечных патологий. По статистическим данной мозжечковой атаксией Пьера-Мари страдают 1 человек на 200 тыс. населения.
Генетическое расстройство в детском и подростковом возрасте протекает бессимптомно и проявляется начиная с третьего десятка жизни.
Причины и течение атаксии Пьера-Мари
Поражение функций мозжечка обусловлено генетической патологией по аутосомно-доминантному типу наследования. Для развития атаксии достаточно генетического нарушения, унаследованного от одного родителя.
Мозжечок — основной координаторный центр, выполняющий двигательные задачи. Полушария его отвечают за согласованность движений, а червь мозжечка — за устойчивость и равновесие.
Патологоанатомические признаки болезни выражены гипоплазией мозжечка, уменьшением нижних олив и истощением варолиева моста. На фоне этого, как правило, происходит дегенерация спиноцеребральных путей, разрушение клеток коры мозжечка и ядер, дегенеративные нарушения продолговатого мозга и в ядрах моста мозга.
В зависимости от сосредоточения мозжечкового поражения атаксию разделяют на динамическую и статико-локомоторную. В первом случае патологические нарушения обнаруживаются в полушариях, что обуславливает десинхронизацию мышечных ритмов (дисметрия, скандированная речь, непроизвольное дрожание туловища, головы, конечностей и др.) При статико-локомоторной форме затронут червь, что вызывает расстройство походки, устойчивости и равновесия.
Несмотря на врожденный характер атаксия Пьера-Мари манифестирует, начиная с 20 лет и старше. Провоцирующими факторами выступают инфекционные болезни (сальмонеллез, зоонозная инфекция, бактериальная пневмония, брюшной или сыпной тиф, пиелонефрит, менингит и пр.). В качестве экзогенных причин могут послужить черепно-мозговая травма, перелом тазовых костей или грудной клетки, глубокие ожоги и интоксикация различной природы.
Наследственная мозжечковая патология отличается непрерывно прогрессирующими проявлениями. Симптоматическая терапия не обеспечивает периодов ремиссии. Внешние патогенные факторы в виде различных болезней ухудшают состояние пациента. В дальнейшем тяжелое состояние проходит и возвращается типичный симптомокомплекс мозжечковой патологии.
Симптомы атаксии Пьера-Мари
Основным признаком наследственной болезни будут нервно-мышечные нарушения моторики, которые не ограничиваются отдельной группой мышц или конкретными движениями.
Мозжечковую атаксию отличают характерные симптомы:
- нарушение походки;
- расстройство статики;
- тремор конечностей и тела;
- мышечные подергивания;
- непроизвольные частые колебательные движения глаз;
- медленная речь;
- изменение почерка в сторону значительного увеличения букв;
- снижение мышечного тонуса.
Атаксия начинает развиваться с нарушения в походке: больной передвигается покачиваясь. Иногда первыми симптомами будут прострелы в поясничной области. Затем патология затрагивает руки, отмечается их дрожание.
При болезни Пьера-Мари можно наблюдать парез конечностей, на фоне которого повышены сухожильные рефлексы. Нередко у пациента фиксируют сгибательные и разгибательные пирамидные рефлексы стоп. Достаточно часто встречаются церебральные симптомы: опущение верхнего века (птоз), затрудненная конвергация глаз, атрофия зрительно нерва.
У 50% больных наблюдаются психические и умственные нарушения: деменция, олигофрения, депрессия.
Дифференциальная диагностика атаксии Пьера-Мари
Немаловажное значение в диагностике имеет скрупулёзный сбор сведений генетической заболеваемости ближайших родственников и особенностей клинической картины.
Диагностика предполагает проведение лабораторных и инструментальных исследований:
- Электроэнцефалография (ЭЭГ). Обнаруживает диффузную дельта/тета-активность и ослабление альфа-ритма;
- Электромиография. Выявляет аксонально-демиелинизирующее нарушение волокон периферических нервов;
- Магнитно-резонансная томография. Фиксирует морфологические изменения структур спинного и головного мозга;
- ДНК-тест. Определяет генетическую природу атаксии;
- Лабораторные анализы. Позволяют распознать нарушение обмена аминокислот.
Единичный случай в семье мозжечковой атаксии требует более глубокого обследования и дифференциальной диагностики. Помимо вышеперечисленных заболеваний, имеющих симптомокомплекс атаксии, проводят освидетельствование на предмет исключения новообразования мозжечка, абсцесса или гематомы головного мозга, церебеллита и гидроцефалии.
При офтальмологических расстройствах требуется обследование у соответствующего специалиста.
Для подтверждения предварительного диагноза семейной атаксии необходима консультация генетика.
Диагностика атаксии Пьера-Мари
Симптомокомплекс мозжечковой атаксии идентичен клинической картине наследственной атаксии Фридрейха. Поэтому при постановке диагноза возникают трудности.
Основное отличие — тип наследования. Доминантное наследование характерно для мозжечкового заболевания Пьера-Мари. Рецессивный вид свойственен атаксии Фридрейха. Учитывается возраст, в котором проявились симптомы заболевания. Более ранняя манифестация свойственна аутосомно-рецессивному характеру болезни.
Невролог исследует изменения сухожильных рефлексов, которые увеличены при мозжечковой форме атаксии и понижены при болезни Фридрейха. Кроме того, атаксии Пьера-Мари не характерны костные деформации и потеря чувствительности.
Весьма затруднительно дифференцировать рассеянный склероз и мозжечковую атаксию. Для обоих заболеваний присущи пирамидные дефекты стоп, глазодвигательные расстройства, а также нервно-мышечные нарушения моторики. Однако при рассеянном склерозе в противовес атаксии возможны периоды ремиссии. Кроме того, отличительной чертой склероза являются глубокий парапарез и более выраженные тазовые нарушения.
Лечение и прогноз атаксии Пьера-Мари
Ведущим врачом в данном случае является невропатолог. Он разрабатывает схему консервативной терапии, которая направлена на нивелирование симптоматики и включает в себя:
- Общеукрепляющий медикаментозный комплекс. Назначаются препараты, подавляющие фермент холинэстераза (дезагреганты), предупреждающие повреждение нейронов мозга (нейропротекторы), витамины группы РР, В и С;
- Лечебная физкультура, кинезиотерапия — основные реабилитационные мероприятия. Задача тренировок — лечение движением, укрепление мышц и ослабление симптома дискоординации. При статистической мозжечковой атаксии подбираются упражнения для тренировки равновесия. Для динамической атаксии разрабатывается тренировочный комплекс, повышающий согласованность и точность движений.
- Физиотерапия. Проводится с целью предупреждения контрактуры конечностей, атрофии мышц, коррекции походки, улучшения координации, поддержки общефизической формы;
- Массаж, мануальная- и рефлексотерапия. Проводится для улучшения обменных процессов.
Прогноз наследственной атаксии Пьера-Мари неблагоприятный для трудовой деятельности. Симптомы прогрессируют на протяжении жизни, трудоспособность снижается, а психические нарушения усугубляются. Больной инвалидизируется.
Тем не менее при условии постоянного выполнения симптоматической терапии и соблюдения щадящего режима, прогноз для жизни хороший.
Наследственная мозжечковая атаксия Пьера-Мари — наследственно предопределенная деформация полушарий и червя мозжечка, с демиелинизацией мозжечкового вещества. Постоянно прогрессирующее и характеризующееся дегенеративными трансформациями. Начинает формироваться после 20 лет. Симптомам заболевания сопутствуют зрительные нарушения, гиперрефлексия и значительное снижение интеллекта.
Диагноз устанавливается на базе результатов неврологического и офтальмологического освидетельствования, МРТ головного мозга, УЗДГ или МРА мозговых сосудов, после консультации специалиста — генетика. Лечение является симптоматическим, включает назначение успокоительных средств, антидепрессантов, миорелаксантов и ноотропов. Абсолютно вылечить заболевание невозможно. Пациентам рекомендована лечебная физкультура, гидротерапия, витамины.
Общая информация
Впервые заболевание очень детально изложил Пьер Мари в 1893г. Ученый выделил заболевание в особенную форму и определил различные от атаксии Фридрейха проявления. Атаксии Пьера-Мари присущи другие отличительные клинические симптомы, иной способ наследования, более старший возраст проявления. Заболевание обнаруживается у людей 20-45 лет, независимо от пола. Наблюдается 1 случай болезни на 200 000 человек. Патологические трансформации в структуре при мозжечковой атаксии располагаются в веществе ядер и коры мозжечка. Спинномозговые пути и боковые ответвления спинного мозга выражены не ярко. Дегенеративные преобразования могут поражать ядра моста продолговатого мозга.
Этиология и течение заболевания
Наследственная мозжечковая атаксия воспроизводится по наследству аутосомно-доминантным способом, развивается при получении патологического гена от одного из родителей. Внешние причины оказывают содействие развитию заболевания и значительно отягощают течение. Причинами могут стать разнообразные инфекции (брюшной тиф, бруцеллез, дизентерия, пиелонефрит, пневмония), различные травмы мозга и костей скелета, интоксикации, беременность.
Симптоматика проявляется безостановочно и прогрессирует неуклонно без выраженных промежутков обострения и затихания. Атаксия Пьера-Мари может видоизменять клинические проявления под влиянием внешних факторов или после перенесенных инфекций. Изменчивость клиники значительно затрудняет установление заключения. После инфекционного заболевания возможно наступление резкого обострения, а позже — частичное восстановление, которое ошибочно принимается за улучшение.
Клинические признаки
Классические клинические случаи не обходятся без глазодвигательных нарушений и снижения функций зрения. Постепенное омертвение зрительных нервов приводит к сужению полей зрения. Все эти признаки неуклонно прогрессируют. Регистрируется опущение верхнего века. Больной поднимает брови в попытке уменьшить птоз — это придает лицу удивленный вид. Нередко болезнь выражается косоглазием, нарушением бинокулярного зрения, к которым приводит парез отводящего нерва.
К перечисленным нарушениям понемногу присоединяются психические недомогания. Течение заболевания усугубляется снижением умственных процессов и памяти, развивается депрессивное невротическое состояние. При тяжелых клинических случаях бывают эпилептические приступы.
Дифференциальное диагностирование
Заболевание имеет мозжечковый характер и требует неврологической дифференциации от других видов атаксий — сенситивной и вестибулярной. Гипорефлексия, присущая атаксии Фридрейха. Атаксия Пьера-Мари имеет в неврологической клинике имеет нарастание сухожильных рефлексов, мышечный гипертонус. Имманентны спастические судороги в нижних конечностях, подергивания ступней. При унаследованной мозжечковой атаксии не обнаруживаются деформации костей скелета, которые присущи болезни Фридрейха.
Клинические выражения рассеянного склероза очень сходны с симптомами наследственной мозжечковой атаксии. Для болезни свойственно неуклонное возрастание клинических проявлений без периодов ремиссии. Трудности постановки диагноза возникают во время или после инфекционных заболеваний, так как меняется характер течения основного недуга. В таком клиническом случае необходим детальный и точный сбор анамнеза с целью выявления признаков рассеянного склероза — парапареза нижней части тела, сопровождающегося патологическими рефлексами и гиперрефлексией, императивными тазовыми нарушениями, снижением брюшных рефлексов, с височной стороны наблюдается изменение цвета дисков зрительных нервов.
Диагностика
Невролог делает заключение об атаксии Пьера-Мари без особых затруднений при наличии классической клинической картины и случаев болезни в нескольких поколениях. Случайные, эпизодические заболевания нуждаются в детальном обследовании больного, скрупулезного обследования больного и проведения дифференциальной диагностики с другими видами атаксий, рассеянным склерозом, нейросифилисом.
Обязательно проведение компьютерной томографии, магнитно-резонансной томографии, ревизию спинных сосудов, МРА, УЗДГ с целью исключения опухолей мозжечка, вторичных органических патологий, васкулярных расстройств.
Офтальмолог диагностирует зрительные расстройства. Обязательна проверка качества зрения, исследование бинокулярного зрения. Обязательно проведение офтальмоскопии, периметрии, распознавание угла косоглазия.
Желательна консультация врача-генетика для точного определения заболевания.
Лечение и прогноз
Терапия имеет постоянный, комплексный характер. В лечении принимают участие неврологи, офтальмологи и психиатры. Полное излечение от заболевания невозможно. Лечение носит симптоматическую природу. В основе терапевтических мероприятий лежит назначение антидепрессантов (флуоксетин, тетриндол, амитриптилин, циталопрам), успокоительных средств (настойка пиона, валериана, магния сульфат). Обязательно применять медпрепараты для стимулирования умственного функционирования и высших психических функций — пирацетам, гамма-аминомасляная кислота, медпрепараты гинго-билоба. Кроме этого назначаются средства, снижающие тонус скелетной мускулатуры — баклофен, мелликтин, кондельфин.

Рекомендованы витаминные комплексы с непременным присутствием витаминов группы В, РР, С. Обязательно санаторно-курортное лечение, водотерапия, лечебная физкультура, соблюдение правильного трудового режима. Важно исключить физические нагрузки и психические потрясения, как отрицательные, так и положительные.
Комплексное лечение оптимизирует общее состояние больного и дает возможность сохранить нормальную жизнь в течение долгого времени, насколько это представляется возможным, когда речь идет об унаследованной мозжечковой атаксии.
Прогноз выздоровления при наследственной мозжечковой атаксии крайне неблагополучный. На протяжении заболевания симптомы обостряются и неизменно приводят к инвалидизации. Тяжелое протекание болезни приводит к тому, что больной становится прикованным к постели. Смерть наступает от миокардита или сердечной недостаточности. В лучшем варианте при хорошем присмотре больной может дожить до 40-50 лет.

Наследственная мозжечковая атаксия Пьера-Мари — генетически детерминированное неуклонно прогрессирующее поражение мозжечка, связанное с его дегенеративными изменениями. Развивается после 20 лет. В клинической картине мозжечковая атаксия сочетается с гиперрефлексией, офтальмологическими расстройствами и снижением интеллекта. Диагностический алгоритм предусматривает неврологический и офтальмологический осмотр, МРТ головного мозга, УЗДГ или МРА церебральных сосудов, генетическое консультирование. Радикальная терапия не разработана, осуществляется симптоматическое лечение антидепрессантами, миорелаксантами, седативными и ноотропами. Рекомендуется ЛФК, витаминотерапия и водолечение.

- Причины и течение атаксии Пьера-Мари
- Симптомы атаксии Пьера-Мари
- Дифференциальная диагностика атаксии Пьера-Мари
- Диагностика атаксии Пьера-Мари
- Лечение и прогноз атаксии Пьера-Мари
- Цены на лечение
Общие сведения
Наследственная мозжечковая атаксия была подробно описана Пьером Мари в 1893г. как отличающаяся от известной на то время атаксии Фридрейха нозологическая форма. Действительно, атаксия Пьера-Мари имеет другой тип наследования, более старший возраст манифестации и свои клинические отличия. В связи с этим, несмотря на некоторую общность морфологического субстрата этих заболеваний в виде дегенерации тканей мозжечка и его проводящих путей, в современной неврологии окончательно утвердилось их выделение в самостоятельные нозологические единицы.
Как правило, атаксия Пьера-Мари манифестирует в возрасте от 20 до 45 лет. Мужчины и женщины заболевают одинаково часто. Распространенность патологии составляет 1 случай на 200 тыс. человек. Морфологические изменения при наследственной мозжечковой атаксии преобладают в тканях ядер и коры мозжечка, менее выражено поражение спиноцеребеллярных путей и боковых канатиков спинного мозга, процессы дегенерации могут наблюдаться в ядрах моста и продолговатого мозга.
Причины и течение атаксии Пьера-Мари
Наследственная мозжечковая атаксия имеет доминантный механизм наследования, т. е. развивается при получении дефектного гена от одного из родителей. Зачастую экзогенные причины выступают в роли триггеров, провоцирующих начало заболевания и усугубляющих его течение. К подобным факторам относятся: различные инфекции (брюшной тиф, сыпной тиф, бруцеллез, сальмонеллез, дизентерия, иерсиниоз, бактериальная пневмония, пиелонефрит и др.), беременность, травмы (ЧМТ, повреждения грудной клетки, переломы таза, сильные ожоги), интоксикации.
Атаксия Пьера-Мари характеризуется постоянным и неуклонно прогрессирующим нарастанием патологической симптоматики. Выделение периодов обострения заболевания и его ремиссии не наблюдается. Под влиянием перенесенных инфекционных заболеваний и прочих экзогенных воздействий атаксия Пьера-Мари может изменить характер своего течения, что значительно затрудняет ее диагностику. Так, после перенесенной инфекции наблюдается резкое ухудшение состояния пациента с последующим частичным восстановлением, имитирующим улучшение болезни.
Симптомы атаксии Пьера-Мари
Типичные случаи заболевания сопровождаются расстройством зрения и глазодвигательными нарушениями. Первые включают сужение зрительных полей и падение остроты зрения, обусловленные постепенно прогрессирующей атрофией зрительного нерва. Вторые представлены неполным птозом, недостаточность конвергенции, косоглазием из-за пареза отводящего нерва. Возможны нистагмоидные подергивания глаз. Пытаясь уменьшить явление птоза, пациенты поднимают брови, что придает их лицу удивленное выражение. Дополняют клинику расстройства психики, снижение мыслительных функций и памяти, развитие депрессивного невроза.
Дифференциальная диагностика атаксии Пьера-Мари
Неврологическое обследование позволяет исключить прочие виды атаксии (вестибулярную, сенситивную) и установить ее мозжечковый характер. В отличие от атаксии Фридрейха, характеризующейся гипорефлексией и снижением мышечного тонуса, в неврологическом статусе больных при атаксии Пьера-Мари отмечается повышение сухожильных рефлексов и мышечная гипертония. Особенно типичен спастический тонус в ногах, вызывается клонус стоп. Обычные для болезни Фридрейха выраженные деформации скелета отсутствуют.
Весьма сходной может быть симптоматика наследственной мозжечковой атаксии и рассеянного склероза. Отличительной особенностью первой является постепенное неуклонное прогрессирование без периодов ремиссии, однако различные инфекционные заболевания и травмы могут изменять характер ее течения, вызывая значительные затруднения в постановке диагноза. В подобных случаях важно тщательное исследование анамнестических данных, выявление типичного для рассеянного склероза симптомокомплекса: более четкой пирамидной симптоматики (обычно нижний парапарез спастического типа со значительной гиперрефлексией и патологическими рефлексами) исчезновения брюшных рефлексов, тазовых нарушений (императивные позывы), побледнения дисков зрительных нервов с височной стороны.
Диагностика атаксии Пьера-Мари
При наличии типичной клиники и прослеживании ее в нескольких поколениях диагноз не представляет для невролога особых затруднений. Спорадические случаи заболевания требуют более углубленного обследования пациента и тщательной дифдиагностики с другими видами мозжечковой атаксии, рассеянным склерозом, нейросифилисом.
При необходимости проводят исключение приобретенной органической патологии: опухолей мозжечка (медуллобластомы, астроцитомы, гемангиобластомы), церебеллита, сосудистых нарушений, сдавлений мозжечка при гематомах или абсцессах головного мозга, окклюзионной гидроцефалии. Для этого используют КТ и МРТ головного мозга, УЗДГ и МРА церебральных сосудов.
Диагностику офтальмологических расстройств проводит офтальмолог. Обследование включает проверку остроты зрения, исследование конвергенции, офтальмоскопию, периметрию, измерение угла косоглазия и пр. Для более точной верификации диагноза может потребоваться консультация генетика.
Лечение и прогноз атаксии Пьера-Мари
В лечении пациентов наряду с неврологами принимают участие офтальмологи и психиатры. Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение. В основном это антидепрессанты (амитриптилин, флуоксетин, циталопрам, тетриндол), седативные средства (валериана, пион, магния сульфат), ноотропы (гамма-аминомасляная кислота, пирацетам, экстракты гинго-билоба), препараты, уменьшающие мышечный тонус (меликтин, баклофен, кондельфин). Рекомендованы витамины гр. В, вит. РР и вит. С; бальнеотерапия, лечебная физкультура. Не маловажное значение имеет соблюдение правильного рабочего режима, исключение физических и психологических перегрузок.
Прогноз относительно выздоровления неблагоприятный. Симптомы заболевания постоянно усугубляются и приводят к инвалидизации. Однако систематическое применение симптоматического лечения, регулярное выполнение специального комплекса ЛФК и других рекомендаций делают благоприятным прогноз для жизни пациента.
Читайте также: