
Наследственный семейный амилоидоз без невропатии
Наследственный амилоидоз – группа заболеваний, которые характеризуются накоплением нерастворимых белковых комплексов (амилоида) в различных тканях – главным образом, в нервной и пищеварительной системах, почках и миокарде. Симптомами являются неврологические нарушения, патологии сердца (кардиомиопатия), нефропатия и диспепсические расстройства. При различных формах наследственного амилоидоза превалирует поражение определенной системы. Диагностика производится на основании клинической картины и гистологического изучения тканей, для некоторых форм разработаны методы молекулярно-генетического определения. Лечение включает использование иммуносупрессивных препаратов, поддерживающую и симптоматическую терапию.
- Причины наследственного амилоидоза
- Классификация и симптомы наследственного амилоидоза
- Диагностика и лечение наследственного амилоидоза
- Прогноз наследственного амилоидоза
- Цены на лечение
Общие сведения
Наследственный или семейный амилоидоз – генетически обусловленное нарушение обмена протеинов, при котором происходит образование аномального белка с его последующим осаждением в составе иммунных комплексов в различных тканях. Как нозологическая единица амилоидоз известен давно, но генетическая природа некоторых форм этого состояния была обнаружена только в последние десятилетия. Дифференцировка наследственных и ненаследственных форм значительно осложняется тем, что ряд генетически обусловленных патологий (например, периодическая болезнь) также характеризуются отложением амилоида, но сейчас принято считать, что белковые нарушения при таких заболеваниях имеют вторичный характер.
В настоящий момент к первичному наследственному амилоидозу достоверно относят только типы патологии, обусловленные мутацией гена TTR. Дискуссионным вопросом является классификация так называемого амилоидоза финского типа – установлено, что это заболевание обусловлено дефектом гена GSN, однако до сих пор неясно, являются ли отложения амилоида при этом первичным нарушением или же следствием каких-либо иных процессов. Некоторые врачи-генетики ассоциируют этот тип заболевания с наследственным амилоидозом 4-го типа, но имеющим другую генетическую природу. Встречаемость наследственного амилоидоза не определена по причине редкости этого состояния.
Причины наследственного амилоидоза
Непосредственной причиной первичного наследственного амилоидоза являются дефекты в гене TTR, который располагается на 18-й хромосоме. В настоящий момент идентифицировано более 80-ти разновидностей мутаций этого гена, многие из которых приводят к разнообразным патологиям – помимо всех форм наследственного амилоидоза, к ним относят дистранстиретинемическую гипертироксинемию и семейные формы запястного туннельного синдрома. Практически все дефекты TTR наследуются по аутосомно-доминантному механизму, продуктом экспрессии данного гена является белок транстиретин, выполняющий функции транспортного протеина для тироксина и ретинола. Известны и другие патологии, которые вызваны различными повреждениями структуры и образования транстиретина – среди них системный старческий амилоидоз и амилоидная полинейропатия.
Патогенез наследственного амилоидоза связан с нарушением стабильности молекул транстиретина – по причине мутации гена TTR изменяется структура этой белковой молекулы и она становится нестабильной. В ряде случаев нестабильность незначительно влияет на ее функции, но иногда конформация молекулы изменяется настолько, что она приобретает иммуногенные свойства. Аномальный протеин вызывает реакцию, сходную с аллергической (иммунокомплексный тип), в результате чего возникают нерастворимые белковые комплексы, склонные к отложению в экстрацеллюлярном пространстве тканей. Клинические проявления наследственного амилоидоза зависят от того, где преимущественно происходит отложение протеиновых комплексов, которые вызывают нарушения функциональности органов.
Классификация и симптомы наследственного амилоидоза
Наследственный амилоидоз, обусловленный мутациями гена TTR, способен проявляться нарушениями со стороны разных органов и тканей – нервов, желудка и 12-типерстной кишки, почек, сердца, глаз. При этом клиническая картина заболевания зависит от характера мутации, различные формы отличаются между собой превалирующим поражением каких-либо органов, выраженностью нарушений и длительностью течения. На сегодняшний день выделяют 6 основных фенотипических форм наследственного амилоидоза.
Тип 1 (синдром Андраде) – является наиболее распространенным вариантом наследственного амилоидоза, впервые был описан в 1952-м году. Наиболее распространен в Португалии, а также в бывших португальских колониях (Латинской Америке). Начало этой формы наследственного амилоидоза чаще всего регистрируется в возрасте 30-40 лет, первым симптомом является нарушение чувствительности (болевой, температурной, тактильной) на коже нижних конечностей. Полиневропатия постепенно становится все более выраженной, помимо поражения чувствительных нервов со временем присоединяются двигательные расстройства мышц дистальных отделов ног. Из-за нарушения иннервации наследственный амилоидоз часто приводит к трофическим язвам нижних конечностей. Патология затрагивает и вегетативную нервную систему – у больных наблюдаются нарушения потенции и ортостатическая гипотония. На конечных этапах развития наследственного амилоидоза 1-го типа отложения белка возникают в почках, сердце и роговице, вызывая нарушения в этих органах и приводя к летальному исходу – чаще всего, через 10-13 лет после появления первых симптомов.
Тип 2 (синдром Рукавина) – очень редкий тип наследственного амилоидоза, который достоверно был описан лишь у представителей двух семей в США в 1956-м году. Он проявляется в 35-45 лет и начинается с выраженного туннельного синдрома запястного канала, к которому через несколько лет присоединяются поражение чувствительных и вегетативных нервов. Этот тип наследственного амилоидоза прогрессирует очень медленно, через 15-20 лет после начала заболевания могут отмечаться отложения амилоида в кишечнике, сердце, почках.
Тип 3 (синдром Ван Аллена) – разновидность наследственного амилоидоза (наследственной амилоидной невропатии), характеризующаяся превалирующим поражением нервов и кишечника. Клинические проявления этого заболевания очень схожи с 1-м типом, основное отличие заключается в отсутствии значительного поражения вегетативной нервной системы. Также у больных этой формой наследственного амилоидоза часто отмечаются гастриты и язвы 12-типерстной кишки.
Тип 4 или наследственный амилоидоз финского типа был впервые описан в 1971-м году, по поводу его генетической природы, как уже было сказано выше, в настоящий момент ведется активная дискуссия. В основном при этой форме заболевания поражаются черепно-мозговые нервы, нарушается чувствительность верхних конечностей. Со временем возникает решетчатая атрофия роговицы и снижается тургор кожи, на заключительных этапах развития патологии возникают выраженная полинейропатия и отложения амилоида в сердце. В отличие от других форм наследственного амилоидоза, этот тип может наследоваться по аутосомно-рецессивному механизму (в том случае, если заболевание обусловлено мутациями гена GSN).
Наследственный кардиомиопатический амилоидоз или наследственный амилоидоз датского типа проявляется слабо выраженными неврологическими нарушениями и значительным поражением миокарда (амилоидоз сердца). Отложения амилоида приводят к быстрому развитию кардиомиопатии и сердечной недостаточности, что становится причиной летального исхода.
Нефропатический амилоидоз с глухотой, крапивницей и лихорадкой – редкая, но достаточно тяжелая форма наследственного амилоидоза, при котором у больных в возрасте 20-30 лет возникают лихорадка неустановленного генеза и частые приступы крапивницы. Затем к этим проявлениям присоединяется нейросенсорная тугоухость, которая в последующем осложняется полной глухотой. Поражение почек (амилоидоз почек) нередко приводит к хронической почечной недостаточности, что и становится причиной смерти от уремии большинства больных.
Диагностика и лечение наследственного амилоидоза
Диагностика наследственного амилоидоза производится на основании данных осмотра пациента, общеклинических анализов, оценки состояния ключевых органов (нервной системы, сердца, почек), гистологического изучения тканей, молекулярно-генетических исследований. При осмотре могут выявляться признаки неврологических поражений (нарушения чувствительности и двигательных функций, гипо- или арефлексия), в запущенных случаях отмечается атрофия мышц, обусловленная отсутствием иннервации. Офтальмологическое обследование при наследственном амилоидозе нередко обнаруживает признаки отложения амилоида в стекловидном теле, возможна атрофия или изъязвление роговицы. В ряде случаев могут отмечаться нарушения со стороны кожных покровов – снижение тургора, крапивница, трофические язвы.
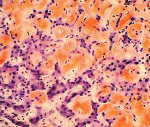
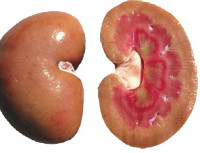
Ультразвуковое исследование сердца и почек при наследственном амилоидозе также нередко дает немало информации об этом заболевании. В случае отложения амилоида в почках наблюдается их увеличение и уплотнение структуры, иногда определяется наличие кист. На конечных этапах развития амилоидоза может возникать уменьшение почек по причине их атрофии. На ЭКГ уменьшается амплитуда и вольтаж всех зубцов, ЭхоКГ обнаруживает значительное увеличение толщины миокарда и размеров левого желудочка. В ряде случаев методом УЗИ при наследственном амилоидозе можно выявить отложения амилоида и в других органах – печени, селезенке, крупных сосудах. При биопсии пораженных органов больных, страдающих наследственным амилоидозом, отмечается типичная для этого заболевания гистологическая картина – отложение эозинофильных масс с концентрацией вокруг кровеносных сосудов. Современная генетика способна выявлять мутации в гене TTR методом прямого секвенирования.
Специфического лечения наследственного амилоидоза не существует, в основном применяется симптоматическая и поддерживающая терапия. Для замедления течения заболевания используют иммуносупрессивные средства (в частности – кортикостероиды) изредка назначают цитотоксические препараты. В случае значительного поражения почек по причине наследственного амилоидоза показан гемодиализ. Симптоматическое лечение также необходимо при кардиомиопатии и сердечной недостаточности. Рекомендуется принимать повышенные дозировки витаминов группы В, так как они замедляют развитие нейропатии и способствуют улучшению общего состояния больного.
В большинстве случаев прогноз наследственного амилоидоза неблагоприятный, так как многие формы этого заболевания приводят к летальному исходу через 10-25 лет после его начала. Симптоматическая и поддерживающая терапия, гемодиализ, применение иммуносупрессоров способны замедлить развитие этой патологии. С учетом того, что чаще всего симптомы наследственного амилоидоза проявляются в 30-40 лет, своевременная диагностика заболевания и разумная поддерживающая терапия способны обеспечить выживаемость больных до старости без значительного ухудшения качества жизни.
OMIM 105210
Наша команда профессионалов ответит на ваши вопросы
Наследственные амилоидозы это клинически и генетически гетерогенная группа аутосомно-доминантных наследственных заболеваний, характеризующихся отложением нерастворимых белковых фибрилл в эксатрацеллюлярном матриксе. Симптомы пациентов с транстиретиновым амилоидозом, как правило, следующие: полинейропатия, туннельный синдром запястного канала, недостаточность вегетативной нейрорегуляции, кардиомиопатия, расстройство функций желудочно-кишечной системы, почечная недостаточность, помутнение стекловидного тела. На поздних стадиях заболевания сильная диарея с мальабсорбцией, кахексия, инвалидизирующая нейропатия, расстройство сердечной деятельности, и выраженная ортостатическая гипотензия, доминирующая в клинической картине. Смерть обычно наступает через 5-15 лет после появления симптомов.
Среди других проявлений амилоидоза следует отметить поражение сердца. Основные проявления амилоидоза сердца – нарушения ритма и проводимости, прогрессирующая сердечная недостаточность, кардиалгии, обусловленные, по-видимому, поражением мелких коронарных артерий. На ЭКГ обнаруживают снижение вольтажа зубцов, инфарктоподобные изменения. При эхокардиографии выявляют резкое утолщение и уплотнение миокарда, уменьшение размера полости левого желудочка. В результате поражения надпочечников при амилоидозе развивается артериальная гипотензия.
Наследственный амилоидоз классифицируется по клинической картине на 4 типа:
Тип I (ANDRADE). Это состояние впервые было описано в северной Португалии Andrade (1952г.), но его также наблюдали в Бразилии, Японии и других местах. Данная форма нейропатического амилоидоза является наиболее распространенной. Заболевание обычно начинается в возрасте 30–40 лет с постепенного снижения болевой и температурной чувствительности в нижних конечностях. При этом часто отмечаются боли, а иногда и парестезии. В конечном итоге появляются нарушения чувствительности в верхних конечностях, а поздние проявления заболевания включают потерю проприоцептивной и вибрационной чувствительности, атрофию и слабость мышц в дистальных отделах конечностей и отсутствие сухожильных рефлексов. Вегетативная недостаточность проявляется в виде импотенции, ортостатической гипотензии, дистального ангидроза, зрачковых нарушений, расстройства функций мочевого пузыря и кишечника, которые часто бывают ранними симптомами болезни. Утрата болевой чувствительности в стопах может вести к возникновению нейрогенной артропатии и изъязвлению кожи. Отложения амилоида могут наблюдаться также в стекловидном теле глаз, почках и сердце. Заболевание медленно прогрессирует, и в общей массе больные умирают приблизительно через 10 лет после начала болезни.
Тип II (RUKAVINA). Этот вариант был описан у двух семей в США в 1956 и 1969гг. Он часто проявляется в виде туннельного синдрома запястного канала, который развивается в среднем возрасте. Вслед за этим появляются симптомы более генерализованной сенсорной и вегетативной невропатии и отложения амилоида во внутренних органах. Заболевание носит медленно прогрессирующий характер.
Тип III (VAN ALLEN). Клинические особенности наследственной амилоидной невропатии типа III аналогичны тем, что наблюдаются при типе I, но вегетативные нарушения выражены менее ярко. Среди больных отмечается высокая частота возникновения язвы двенадцатиперстной кишки.
Тип IV (MERETOJA). Это состояние характеризуется невропатией черепных нервов, поражением верхних конечностей, в основном кистей, решетчатой дистрофией роговицы и снижением тургора кожи. Болезнь обычно начинается на третьем десятилетии жизни с появления помутнений в роговице. Признак невропатии в конечностях присоединяются позже.
Впервые заболевание было описано в Финляндии Meretoja и Терро (1971). Известны также наследственный кардиопатический амилоидоз(датский тип), нефропатический амилоидоз с глухотой, лихорадкой и крапивницей.
За наследственный амилоидоз ответственны мутации в гене TTR, расположенном на длинном плече хромосомы 18 и состоит из 127 амино-кислотных остатков.
Ген TTR кодирует белок транстиретин.
Транстиретин — белок, обеспечивающий транспорт тироксина и ретинола. Транспорт ретинола происходит при соединении транстиретина с ретинол-связывающим белком. Транстиретин связан с такими заболеваниями, как старческий системный амилоидоз, семейная амилоидная полинейропатия, семейная амилоидная кардиомиопатия.
Обнаружено более 80 мутаций гена TTR, вызывающих заболевания. Большинство этих мутаций приводит к амилоидозу. Амилоидогенные мутации снижают стабильность белка.
В Центре Молекулярной Генетики проводится анализ частых мутаций Val30Met и Val122Ile гена TTR, а также поиск мутаций во всех экзонах гена TTR.
Нарушение здоровья, относящееся к группе нарушения обмена веществ
51 111 людям подтвержден диагноз Наследственный семейный амилоидоз без невропатии
17 695 умерло с диагнозом Наследственный семейный амилоидоз без невропатии
34.62 % смертность при заболевании Наследственный семейный амилоидоз без невропатии
Заполните форму для подбора врача
Мы свяжемся с вами сразу, как найдем подходящего специалиста
Диагноз Наследственный семейный амилоидоз без невропатии ставится мужчинам на 12.93% чаще чем женщинам
27 107
мужчин имеют диагноз Наследственный семейный амилоидоз без невропатии. Для 9 372 из них этот диагноз смертелен
смертность у мужчин при заболевании Наследственный семейный амилоидоз без невропатии
24 004
женщин имеют диагноз Наследственный семейный амилоидоз без невропатии Для 8 323 из них этот диагноз смертелен.
смертность у женщин при заболевании Наследственный семейный амилоидоз без невропатии
Группа риска при заболевании Наследственный семейный амилоидоз без невропатии мужчины в возрасте 70-74 и женщины в возрасте 70-74
Заболевание чаще всего встречается у мужчин в возрасте 70-74
У мужчин заболевание реже всего встречается в возрасте 0-1, 15-19, 95+
У женщин заболевание реже всего встречается в возрасте 0-1, 95+
Заболевание чаще всего встречается у женщин в возрасте 70-74
Особенности заболевания Наследственный семейный амилоидоз без невропатии
Отсутствие или низкая индивидуальная и общественная опасность
* - Медицинская статистика по всей группе заболеваний E85 Амилоидоз
Этиология
Причины нарушения обмена веществ многообразны и не до конца изучены. Патологический процесс может быть вследствие дисфункций щитовидной железы, надпочечников, половых желез, гипофиза. Огромное значение для правильного обмена веществ имеет режим питания и образ жизни человека. Переедание или голодание, тяжелая и низкокалорийная пища влияют на энергетические и накопительные процессы в организме, нарушая общий баланс системы.
Клиническая картина
Характерными симптомами нарушения обмена веществ являются избыточный вес, отечность, нездоровые кожные покровы, ослабленные волосы и ногтевые пластины. Неправильное питание с повышенным количеством потребляемых животных жиров, как правило, ведет к серьезным сбоям в процессе метаболизма.
Диагностика
Стандарта по диагностике заболевания Наследственный семейный амилоидоз без невропатии не установлено
Диагноз Наследственный семейный амилоидоз без невропатии на 73 месте по частоте заболеваний в рубрике НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВ
Чаще всего встречаются:
Заболевание Наследственный семейный амилоидоз без невропатии на 75 месте по опасности заболеваний в рубрике НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВ
Диагноз ставится на основании жалоб больного и совокупности клинических симптомов. Также используются лабораторные методы диагностики.
| Медицинская услуга | Средняя цена по стране |
| Назначение диетической терапии при оказании паллиативной помощи | Нет данных |
| Назначение лекарственных препаратов при заболевании, вызываемом вирусом иммунодефицита человека (ВИЧ-инфекции) | Нет данных |
| Назначение лекарственных препаратов при онкологическом заболевании у взрослых | Нет данных |
| Назначение лекарственных препаратов при специфических заболеваниях водолазов | Нет данных |
| Назначение лечебно-оздоровительного режима при профессиональных заболеваниях | Нет данных |
| Назначение диетической терапии при профессиональных заболеваниях | Нет данных |
| Назначение лекарственных препаратов при профессиональных заболеваниях | Нет данных |
| Назначение лечебно-оздоровительного режима при туберкулезе | Нет данных |
| Назначение диетической терапии при туберкулезе | Нет данных |
| Назначение лекарственных препаратов при туберкулезе | Нет данных |
| Еще услуги |
| Медицинская услуга | Средняя цена по стране |
| Анализ мочи общий | Нет данных |
| Анализ крови биохимический общетерапевтический | Нет данных |
| Общий (клинический) анализ крови развернутый | Нет данных |
| Общий (клинический) анализ крови | Нет данных |
| Комплекс клинико-психологических исследований для определения характера нарушения высших психических функций, эмоций, личности | Нет данных |
| Комплекс клинико-психологических исследований для оценки факторов риска, и адаптивных ресурсов психики больного | Нет данных |
| Психологическое (психотерапевтическое) консультирование по коррекции факторов риска развития неинфекционных заболеваний повторное | Нет данных |
| Психологическое (психотерапевтическое) консультирование по коррекции факторов риска развития неинфекционных заболеваний первичное | Нет данных |
| Комплекс исследований для оценки возможностей прижизненного родственного донорства гемопоэтических стволовых клеток | Нет данных |
| Комплекс исследований для диагностики нарушений функции надпочечников | Нет данных |
| Еще услуги |
* - Медицинская статистика по всей группе заболеваний E85 Амилоидоз
Лечение
Стандарта по лечению заболевания Наследственный семейный амилоидоз без невропатии не установлено
10 дней требуется врачам на лечение в стационаре
2 часа требуется на курс амбулаторного лечения
0 медицинских процерур предусмотренно при лечении заболевания Наследственный семейный амилоидоз без невропатии
Лечение нарушения обмена веществ – ответственный и зачастую очень сложный процесс. Генетически обусловленные метаболические заболевания требуют постоянного медицинского наблюдения и регулярной терапии. Приобретенные заболевания, как правило, можно остановить на ранних стадиях. При отсутствии своевременного медицинского вмешательства подобные заболевания могут иметь серьезные осложнения.Основное внимание при лечении нарушений обмена следует уделять рациону и режиму питания. Необходимо снизить и в дальнейшем контролировать объем поступающих в пищу животных жиров и углеводов. Частое дробное питание позволяет уменьшить количество пищи, принимаемой единовременно, в результате чего постепенно можно добиться существенного снижения аппетита и уменьшения объема желудка.
Медицинский эксперт статьи

Амилоидоз - нарушение белкового обмена, сопровождающееся образованием в тканях специфического белково-полисахаридного комплекса (амилоида) и поражением многих органов и систем.
Код по МКБ-10
- Е85 Амилоидоз.
- Е85.0 Наследственный семейный амилоидоз без невропатии.
- Е85.1 Невротический наследственный семейный амилоидоз.
- Е85.2 Наследственный семейный амилоидоз неуточнённый.
- Е85.3 Вторичный системный амилоидоз.
- Е85.4 Ограниченный амилоидоз.
- Е85.8 Другие формы амилоидоза.
- Е85.9 Амилоидоз неуточнённый.
Код по МКБ-10
Эпидемиология амилоидоза
Частота заболевания среди мужчин и женщин при первичном амилоидозе одинакова. Возраст начала болезни колеблется от 17 до 60 лет, а длительность болезни - от нескольких месяцев до 23 лет. Сроки начала заболевания трудно установить, так как первые клинические проявления не соответствуют началу отложения амилоида.

[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14]
Причины и патогенез амилоидоза
В зависимости от этиологии и особенностей патогенеза выделяют идиопатический (первичный), приобретённый (вторичный), наследственный (генетический), локальный амилоидоз, амилоидоз при миеломной болезни и APUD-амилоидоз. Наиболее часто встречается вторичный амилоидоз, который по происхождению приближается к неспецифическим (в частности иммунным) реакциям. Он развивается при ревматоидном артрите, анкилозирующем спондилите, туберкулёзе, хроническом остеомиелите, бронхоэктатической болезни, реже при лимфогранулематозе, опухолях почки, лёгкого и других органов, сифилисе, неспецифическом язвенном колите, болезнях Крона и Уиппла, подостром инфекционном эндокардите, псориазе и др.
[15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25]
Симптомы амилоидоза
Клинические проявления амилоидоза разнообразны и зависят от локализации амилоидных отложений, их распространённости. Локализованные формы амилоидоза, например амилоидоз кожи, долго протекают бессимптомно, как и старческий амилоидоз, при котором отложения амилоида в мозге, поджелудочной железе, сердце нередко обнаруживают только на вскрытии.
Что беспокоит?
Классификация амилоидоза
Согласно классификации Номенклатурного комитета Международного союза иммунологических обществ (Бюллетень ВОЗ, 1993) выделяют пять форм амилоидоза.
- AL-амилоидоз (А - amyloidosis, амилоидоз, L - light chains, лёгкие цепи) - первичный, связанный с миеломной болезнью (амилоидоз регистрируют в 10-20% случаев миеломной болезни).
- АА-амилоидоз (acquired amyloidosis, приобретённый амилоидоз) - вторичный амилоидоз на фоне хронических воспалительных, ревматических, заболеваниях, а также при средиземноморской семейной лихорадке (периодической болезни).
- ATTR-амилоидоз (А - amyloidosis, амилоидоз, TTR - transthyretin, транстиретин) - наследственно-семейный амилоидоз (семейная амилоидная полинейропатия) и старческий системный амилоидоз.
- Аβ2М-амилоидоз (А - amyloidosis, амилоидоз, β2М - β2-микроглобулин) - амилоидоз у больных, находящихся на плановом гемодиализе.
- Локализованный амилоидоз чаще развивается у людей старческого возраста (AIAPP-амилоидоз - при инсулиннезависимом сахарном диабете, АВ-амилоидоз - при болезни Альцгеймера, AANF-амилоидоз - старческий амилоидоз предсердий).
[26], [27], [28], [29], [30], [31], [32]
Периодическая болезнь (или армянская болезнь, или ереванская болезнь, или наследственный семейный амилоидоз без невропатии, или средиземноморская семейная лихорадка, другие названия: пароксизмальный синдром Джэйнуэя — Мозенталя, периодический перитонит, синдром Рейманна, болезнь Сигала — Маму) — сравнительно редкое генетически обусловленное наследственное (которое обусловлено мутацией гена MEFV 16 хромосомы) аутовоспалительное заболевание проявляющееся периодически рецидивирующим серозитом и относительно частым развитием амилоидоза. Встречается преимущественно у представителей народностей, предки которых жили в бассейне Средиземного моря (вне зависимости от места их нынешнего проживания), особенно у армян (в армянской популяции встречается в 1-2% случаев), евреев (чаще сефардов), арабов, и лишь в 6% случаев у лиц прочих национальностей. Встречаемость ПБ среди евреев-сефардов по разным данным составляет от 1:250 до 1:2000 (частота носительства мутантного гена от 1:16 до 1:8), среди армян — от 1:100 до 1:1000 (частота носительства — от 1:7 до 1:4).
Этиология и патогенез. Причина болезни — наследственное нарушение обмена веществ, которые сопровождаются ростом проницаемости сосудов, развитием соединительной ткани и склонностью к экссудации (отечности). В основе ПБ лежит точечная мутация в гене белка пирина, расположенного в коротком плече 16-й хромосомы (16q) рядом с генами аутосомно-доминантного поликистоза почек и туберозного склероза. Пирин — белок первичных гранул нейтрофилов, активно участвующий в регуляции процессов воспаления. Считается, что пирин стимулирует выработку противовоспалительных медиаторов, позволяет контролировать хемотаксис, стабилизирует мембрану гранулоцитов. Нарушение структуры этого белка, имеющее место при ПБ, приводит к повышению выработки провоспалительных медиаторов в лейкоцитах, активации микротубулярного аппарата и спонтанной дегрануляции первичных гранул лейкоцитов, активации молекул адгезии и усиленному хемотаксису лейкоцитов, результатом чего является воспаление.
Долгое время болезнь протекает бессистемно, не вызывая каких-либо обострений. Однако под влиянием комплекса (полностью не изученного) внутренних и внешних факторов развивается доброкачественная опухоль серозных оболочек.
ПБ протекает в виде приступов, основой которых является спонтанная или спровоцированная дегрануляция нейтрофилов с выбросом медиаторов и развитием асептического воспаления преимущественно на серозных и синовиальных оболочках. В периферической крови повышается количество нейтрофилов и острофазовых белков (СРБ — С-реактивный реактивный белок, SAA — сывороточный белок амилоида А и др.). Раздражение медиаторами воспаления рецепторов приводит к развитию болевого синдрома, а воздействие большого количества эндогенных пирогенов на центр терморегуляции — к развитию лихорадки.
Клиника. ПБ проявляется возникающими через определенные интервалы (дни — недели — месяцы) стереотипными приступами лихорадки. Лихорадке могут сопутствовать болевые синдромы, связанные с развитием неспецифического воспаления в серозных и синовиальных покровах. В зависимости от пенетрантности генов эти синдромы могут быть изолированными или сочетаться, но каждый из них сохраняет свой ритм. Любая атака сопровождается лейкоцитозом, увеличением СОЭ и других воспалительных белков, повышением a- и b-фракции глобулинов, снижением активности миелопероксидазы нейтрофилов. Вне приступа дети чувствуют себя хорошо, лабораторные показатели постепенно нормализуются.
Длительность лихорадочного и абдоминального вариантов ПБ обычно составляет от 1 до 3 дней, реже удлиняется до 1–2 нед. Перитонит, как и суставной синдром, наиболее закономерен для детского возраста.
Торакальный вариант с плевральным синдромом встречается реже — около 40% случаев, изолированно — в 8%, в сочетании с абдоминальным синдромом — в 30%. При торакальном варианте развивается одно-двусторонний плеврит со стерильным выпотом. Длительность этого синдрома — 3–7 дней. Как правило, таким больным ошибочно устанавливается диагноз плеврита или плевропневмонии.
Кожные изменения во время приступа ПБ встречаются в 20–30% случаев. Наиболее типичной является рожеподобная сыпь, но могут встречаться пурпурные высыпания, везикулы, узелки, ангионевротические отеки. Иногда клинически ПБ протекает подобно аллергической реакции вплоть до отека Квинке и крапивницы.
Манифестация заболевания может приходиться на различный возраст. Описаны случаи довольно позднего манифестирования ПБ, после 20–25 лет. По нашим наблюдениям, у большинства больных первый приступ ПБ отмечался в возрасте 2–3 лет (9 пациентов), у 1 — с рождения, у 2 — в 0,5–1,5 года, у 2 — в 4–5 лет, у 1 — в 11–12 лет. Частота и периодичность приступов варьируются у разных больных в широких пределах: от нескольких раз в неделю до 1–2 раз в несколько лет. У большинства больных приступы имеют довольно стабильный ритм. Однако в литературе описаны случаи, когда приступы могли прекратиться на несколько лет или, наоборот, возобновиться после длительного перерыва под воздействием внешних факторов (смена места жительства, женитьба или замужество, рождение ребенка, служба в армии и др.). У наших пациентов периодичность приступов была довольно постоянной: у 1-го — 2 раза в нед, у 4-х — 1 раз в нед, у 5-ти — 1 раз в 2–3 нед, у 2-х — 1 раз в мес, у 1 — 1 раз в 2–3 мес, у 1-го — 1 раз в 6–12 мес.
Характеризуется развитием повторяющихся приступов лихорадочных торакоабдоминальгии, суставного синдрома по типу преходящих моноартритов с кожным эризипелоидом либо поражением осевого скелета, схожего с спондилоартропатией. Формы этой болезни — перитонит (боли в животе — наиболее частые проявления данной патологии), васкулярная, суставная, плевральная, менингиальная, смешанная. Начинается лихорадка и болевые синдромы. Белок-амилоид откладывается в тканях и органах (зачастую — в почках). Атака растягивается на несколько дней, после чего самочувствие пациента улучается. До следующего приступа. Ремиссия составляет около 3-7 дней. При абдоминальном варианте у врачей может возникнуть подозрение на аппендицит, поскольку пациент страдает острым животом. Иногда кажется, что вы имеете дело с непроходимостью тонкой кишки либо холециститом. Впрочем, через 2-4 дня все симптомы загадочным образом исчезают. При лихорадочной разновидности недуга больного резко повышается температура, а течение болезни напоминает малярийную лихорадку. Довольно неприятен суставной вариант, проявляющийся в форме рецидивирующего синовита, поли- и моноартрита, артралгии. Может возникнуть преходящий остеопороз. Клинические проявления могут провоцироваться климатическими факторами, приёмом алкоголя, инфекциям, алиментарными отравлениями. Сезонность проявления зависит от региона. Например, для России это межсезонье — весна-лето и лето-осень. Периодическая болезнь и беременность несовместимы. Во всяком случае, частота припадков у будущих матерей ощутимо снижается.
Клинически у детей на фоне приступов ПБ может развиваться гипохромная железодефицитная анемия, гиперпротеинемия, диспротеинемия с увеличением глобулинов α2, β и γ, отмечается высокое содержание фибриногена и сиалопротеинов. Характерны увеличение и уплотнение печени и селезенки. Изменения в моче поначалу отсутствуют или носят транзиторный характер, однако со временем протеинурия становится постоянной и более выраженной, часто наблюдается микрогематурия и цилиндрурия. Появление постоянной протеинурии характеризует переход во вторую, протеинурическую, стадию.
Через некоторое время от начала манифестации у большинства больных отмечается гепатомегалия, которая, по нашим наблюдениям, может варьироваться от +1 до +5 см. Постепенно развивается и спленомегалия, величина которой у некоторых пациентов достигала +7 см. Однако увеличение печени и селезенки выявляется не у всех больных. Очевидно, эти процессы зависят от частоты и количества перенесенных приступов и развития амилоидоза. Ведущим в патогенезе этой стадии амилоидоза является значительный синтез и повышение в плазме крови концентрации белков-предшественников амилоидоза, т. е. диспротеинемия. В протеинурической стадии амилоид появляется не только в пирамидах, но и в половине клубочков почек в виде небольших отложений в мезангии, отдельных капиллярных петлях, а также артериолах. Отмечается выраженный склероз и амилоидоз стромы, сосудов, пирамид и интермедиарной зоны, что приводит к атрофии многих глубокорасположенных нефронов. Продолжительность этой стадии, как и предыдущей, колеблется от нескольких месяцев до многих лет. По мере нарастания тяжести амилоидоза усугубляются лабораторные показатели выраженной активности процесса: значительная протеинурия и диспротеинемия, гиперфибриногенемия, СРБ, гиперкоагуляция. Дальнейшее отложение амилоида в почечной ткани и нарастающая протеинурия приводят к развитию отечного синдрома, появление которого свидетельствует о переходе заболевания в третью, отечную, стадию.
В отечную (нефротическую) стадию амилоидоза количество амилоида в почках увеличивается. Пораженными оказываются более чем 75% гломерул. Прогрессирует склероз интерстиция и сосудов, в пирамидах и интрамедиарной зоне склероз и амилоидоз имеют выраженный диффузный характер. Клинически эта стадия амилоидоза представлена полным нефротическим синдромом, хотя иногда может наблюдаться неполный (безотечный) нефротический синдром. Протеинурия становится массивной и, как правило, неселективной; нарастают циллиндры. Гематурия бывает редко и, как правило, незначительна. Нарастают гепатоспленомегалия, гипопротеинемия, усиливаются диспротеинемия с дальнейшим повышением уровня α1-, α2-, и γ-глобулинов, гиперфибриногенемия, гиперлипемия. Со временем появляется артериальная гипертензия, нарастает азотемия, прогрессирует почечная недостаточность.
Уремическая (азотемическая) стадия развивается в финале заболевания. В связи с нарастающим амилоидозом и склерозом наблюдаются гибель большинства нефронов, их замещение соединительной тканью, развивается ХПН (хроническая почечная недостаточность). Клиническими особенностями ХПН при амилоидозе, отличающими ее от ХПН вследствие других заболеваний, является сохранение нефротического синдрома с массивной протеинурией, часто определяются большие размеры почек, характерно развитие гипотензии. Часто выражен ДВС-синдром (синдром диссеминированного внутрисосудистого свертывания крови) в виде пурпуры, носовых, желудочных и кишечных кровотечений. Возможны тромбозы почечных сосудов с развитием инфарктов ишемического или геморрагического типа.
Диагностика армянской болезни иногда вызывает затруднения. И все же, характерные черты недуга очертить можно:
- Лихорадка. Сопровождает периоды обострений. Схожа по типологии с малярией и характеризуется внезапным ростом температуры до 40-градусной отметки.
- Перитонит. Наблюдается в 85-95% случаев. При воспалении брюшины пациенты госпитализируются и направляются в хирургические отделения.
- Артрит. Суставная форма заболевания, которая прослеживается в 50-80% случаев.
- Торакальная форма. Сюда относятся всевозможные бронхиты, плевриты, проблемы с дыханием. Наблюдается в 30-60% случаев.
- Комбинированные формы. Увеличение селезенки, поражение лимфоузлов, кожная сыпь (отдаленно напоминает рожу). Изредка — асептический менингит.
Вот на какие критерии нужно обращать внимание при диагностировании:
- Периоды коротких атак. Не имеют связи с провоцирующим фактором, стереотипны.
- Заболевание поражает представителей определенных этнических групп, причем у детей и подростков оно проявляется достаточно рано.
- Аналогичные заболевания у родственников.
- Амилоидоз почек. Специфику лабораторных показателей определить при это крайне сложно.
Периодическую болезнь можно диагностировать, прибегнув к рентгенологическому исследованию пораженных органов брюшной полости. Отложение амилоида после каждой атаки влечет за собой нарастающее поражение почек. Почечная недостаточность (хроническая) проявляется впоследствии у 25-40% пациентов.
Для установления диагноза необходимо молекулярное-генетическое исследование MEFV генного локуса 16-й хромосомной пары. Наличие по одной мутации на каждой из хромосом или одной мутации на одной из хромосом плюс соответсвующая клиничиская картина, подтверждают наличие периодической болезни.
Диагностика периодической болезни и амилоидоза. При типичном течении периодической болезни ее диагностика не представляет трудностей. Наибольшая проблема заключается в незнании большинством врачей этой патологии, что приводит к плохой выявляемости даже при наличии симптомов. Диагностика ПБ основывается на 5 пунктах.
При постановке диагноза ПБ в большинстве случаев у врача возникает настороженность в отношении амилоидоза. Но часто первые подозрения на АА-амилоидоз могут возникнуть у педиатра при лечении больных с нефротическим синдромом, резистентных к стандартной глюкокортикоидной терапии.
Только изучение материалов биопсии с обязательным окрашиванием Конго-красным и поляризационной микроскопией позволяет поставить окончательный диагноз АА-амилоидоза. Помимо этого для диагностики можно использовать специфические антитела к АА-фибриллам. Наиболее достоверной является биопсия почки. Частота выявления АА-амилоидоза в этом случае достигает 90–100%. Чем более распространен процесс, тем больше вероятность выявления АА-амилоида в других местах (желудочно-кишечный тракт (ЖКТ) — слизистая и подслизистая, слизистая десны, прямая кишка, жировая биопсия). Наиболее информативной среди непочечных биопсий является биопсия стенки ЖКТ и прямой кишки, при которой вероятность выявления амилоида составляет 50–70%.
Лечение. Во время приступа — НПВП на 3-5 дней. Основным терапевтическим средством является Колхицин. Дозировка препарата — 1-2 мг в сутки. Он стабилизирует мембрану нейтрофилов. В большинстве случае лекарство на корню пресекает зарождение приступов ПБ, сокращает их выраженность и частоту, предупреждает амилоидоз почек. Изначально лечение недуга было преимущественно симптоматическим. Навыки предупреждения приступов появились у врачей в 1972 году, когда был синтезирован колхицин. Фактически, терапия растягивается на весь остаток жизни. До конца не выяснен механизм действия лекарства. Оно ингибирует простагландины, обладает противовоспалительными качествами, уменьшает проницаемость сосудов.
Колхицин обладает антимитотическим эффектом в отношении амилоидобластов при периодической болезни — макрофагов и стабилизирует мембрану нейтрофилов, препятствуя выбросу пирина. Колхицин назначается пожизненно в дозе 1–2 мг/сут. Он хорошо переносится, иногда возникают диспептические явления, которые не требуют полной отмены препарата. Колхицин в большинстве случаев полностью предотвращает появление приступов ПБ или значительно снижает их частоту и выраженность, предотвращает развитие амилоидоза почек, снижает выраженность его проявлений. При почечной недостаточности дозу снижают исходя из степени снижения клубочковой фильтрации.
Прогноз. Частые приступы могут повлечь за собой временную нетрудоспособность. При стабильном развитии амилоидоза может наступить стадия почечной недостаточности, что неизбежно приведет к инвалидности. Положительный эффект наступит, если лечение начать своевременно. Поэтому диспансерное наблюдение приветствуется и настоятельно рекомендуется. При своевременной постановке диагноза и назначении колхицина прогноз ПБ благоприятен. При отсутствии терапии наибольшую опасность представляет развитие почечного амилоидоза, который, по сути, является единственной причиной смерти больных с ПБ. Анализ заболеваемости у взрослых и детей показывает, что при естественном течении периодической болезни приблизительно у 50% больных терминальная стадия почечной недостаточности развивается через 5 лет от момента появления протеинурии, у 75% — в течение 10 лет.
Читайте также: