
Неврологические проявления болезни фабри
Болезнь Фабри – наследственное заболевание, при котором дефект в структуре генов обуславливает недостаточную активность или отсутствие фермента α-галактозидазы A, накопление в органах промежуточных продуктов липидного обмена. Симптоматика включает боли в конечностях, уменьшение потоотделения, депрессию, быструю утомляемость, почечную и сердечную недостаточность, острые нарушения мозгового кровообращения. Для диагностики используется исследование активности фермента и количества гликосфинголипидов в крови и тканях, секвенирование генетического материала. Лечение основано на ферментозаместительной терапии.
МКБ-10
- Причины болезни Фабри
- Патогенез
- Классификация
- Симптомы болезни Фабри
- Осложнения
- Диагностика
- Лечение болезни Фабри
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Болезнь Фабри получила свое название по фамилии немецкого врача-дерматолога Джона Фабри, который в 1898 году подробно описал симптомы патологии у мальчика 13 лет. В это же время схожий клинический случай был выявлен врачом-дерматологом из Великобритании Вильямом Андерсеном, поэтому заболевание имеет другое распространенное название – болезнь Андерсона-Фабри. Менее известные синонимы – церамидтригексозидоз, диффузная универсальная ангиокератома, наследственный дистонический липидоз. Эпидемиология зависит от расовой и этнической принадлежности, составляет 1 случай на 40-120 тыс. новорожденных. Наиболее высокая распространенность определяется в США.
Причины болезни Фабри
Заболевание обусловлено мутацией гена GLA, который кодирует структуру фермента альфа-галактозидазы А, участвующего в расщеплении гликосфинголипидов. Ген локализован на длинном плече Xq 22.1 хромосомы. Полипептидная структура фермента состоит из 429 остатков аминокислот, наличие и порядок расположения которых определяются 1290 парами оснований гена. В результате научных исследований выявлено 599 вариантов мутаций и полиморфизмов гена, которые изменяют стабильность и активность галактозидазы. Самые распространенные причинные мутации – миссенс и нонсенс, на их долю приходится до 93% случаев болезни.
Механизм наследования патологии определен как рецессивный X-сцепленный, но продолжает изучаться. У гемизиготных мужчин присутствует единственная мутантная X-хромосома. Такой набор представляет классический фенотип заболевания. Больные мужчины способны передать мутацию только дочерям, сыновья остаются здоровыми. Дочери, получившие измененные гены от родителя, являются гетерозиготными и, согласно закону рецессивного наследования, должны оставаться клинически здоровыми – носителями. Вероятность передачи патологического аллеля от матери детям обоих полов составляет 50%.
В ходе клинических исследований установлено, что у большинства женщин с рецессивным мутантным геном болезнь проявляется симптомами умеренной выраженности, имеет позднее начало и медленное прогрессирование. У части пациенток выявляются тяжелые проявления патологии, которые требуют неотложной медицинской помощи. Причины развития тяжелых форм заболевания у гетерозиготных женщин остаются неизвестными. Возможно существование феномена инактивации нормальной X-хромосомы.
Патогенез
Патогенетическая основа болезни – дефицит альфа-галактозы А. В норме этот фермент расщепляет терминальные гликолипиды а-галактозила. При ферментной недостаточности в клеточных лизосомах накапливаются промежуточные продукты обмена – тригексозилцерамид и дигалактозилцерамид. Содержание церамид-тригекосзида значительно увеличивается в эндотелиальных и гладкомышечных клетках сосудов, в эпителиальных и перителиальных клетках многих органов. Молекулы дигалактозил-церамида концентрируются в почках, камерах сердца, ЦНС.
Таким образом, основой патогенеза является нарушение метаболизма мембранных гликосфинголипидов. Их депонирование усиливается при повышенном уровне циркулирующих липидов, поступающих в мембраны методом активного всасывания и диффузии. Основные симптомы заболевания обусловлены скоплением продуктов обмена сфинголипидов в малых и крупных сосудах, включают боль в руках и ногах, приступы лихорадки, пурпурные кожные высыпания. Патологические процессы развиваются постепенно, полиморфная клиническая картина зачастую наблюдается у подростков и детей, у взрослых в большей степени поражается один орган.
Классификация
Поскольку патология является генетической, ее начало приходится еще на внутриутробный период. Из-за относительно медленного прогрессирования симптоматика проявляется гораздо позже – к 6-12 или даже к 30 годам. С учетом времени дебюта заболевания и характера поражения внутренних органов выделяют две формы болезни:
- Классическая. Начинается в детстве и раннем подростничестве. Проявляется полиорганным поражением.
- Атипичная. Симптомы дебютируют в зрелом возрасте. Часто наблюдается изолированное повреждение одного органа: почек, сердца, мозга. У представителей мужского пола признаки более выраженные.
- Женская. Данная форма выделяется в англоязычной литературе. Для нее характерно медленно прогрессирующее течение со слабовыраженной симптоматикой, отсутствие ведущего пораженного органа.
Симптомы болезни Фабри
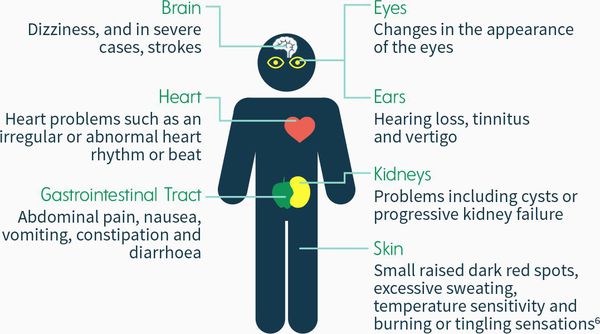
Наиболее частый начальный симптом, который выявляется у 70-80% больных с классическим вариантом патологии – акропарестезии (невропатические боли). Они локализуются в дистальных отделах ступней и ладоней, отличаются высокой интенсивностью и продолжительностью, по характеру – жгучие, колющие, изнуряющие. Бывают хроническими и кризовыми. Хронические боли постоянные, но имеют среднюю интенсивность. Кризы Фабри длятся несколько часов или суток, представляют собой быстро нарастающие болевые приступы, сопровождающиеся гипертермией.
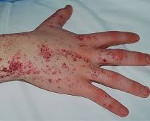
У значительной части больных наблюдается гипогидроз и ангидроз – снижение или полное отсутствие потоотделения. Пациенты плохо переносят жару, часто перегреваются (например, при кризовых болях). Ухудшается работоспособность, снижается толерантность к физическим нагрузкам. Занятия спортом и физический труд провоцируют усиление болей и перегрев тела. Характерный внешний признак болезни – ангиокератомы, мелкие безболезненные папулы красновато-фиолетового цвета, которые располагаются скоплениями на коже. Чаще всего поражается область губ, пальцы рук и ног, гениталии.
Сердечно-сосудистая симптоматика включает аритмии, артериальную гипертензию, гипертрофическую кардиомиопатию. Подростки страдают от приступов гипертензии. Взрослые пациенты испытывают болевые ощущения в области грудной клетки, головокружения, учащенное сердцебиение и одышку, падают в обмороки. Повреждение почек на начальной стадии протекает бессимптомно. При длительном течении болезни снижается фильтрационная и концентрационная функция почек, обращение за врачебной помощью часто происходит уже при хронической почечной недостаточности, требующей проведения гемодиализа.
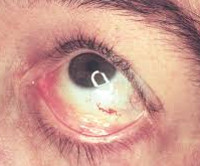
Неврологические симптомы более свойственны взрослым. Типичны головные боли, головокружения. Скопление продуктов метаболизма липидов в сосудах становится причиной нарушения кровообращения в головном мозге. Возникают транзиторные ишемические атаки и инсульты. Офтальмологические расстройства имеют специфические особенности. Наиболее распространенными являются вортексная кератопатия у детей и помутнение хрусталика (двусторонняя передняя и радиальная задняя катаракта) у лиц старшего возраста. Возможно образование конъюнктивальных аневризм, отек сетчатки глаза или папиллоэдема, оптический неврит с выпадением полей зрения, центральными скотомами. Изменения слуха представлены звоном в ушах и ухудшением восприятия звуков. В психоэмоциональной сфере больных преобладают депрессия и тревога.
Осложнения
Нейропатические боли при болезни Фабри с трудом купируются обезболивающими препаратами, поэтому становятся причиной депрессии, низкой мотивации к учебе, профессиональной деятельности и иной социальной активности. В тяжелых случаях акропарестезии провоцируются минимальной физической нагрузкой или умственным переутомлением. В итоге дети и подростки переходят на домашнее обучение, что сопровождается сужением круга друзей, формированием ощущения изолированности. Пациенты находятся в группе риска по совершению суицидальных попыток. Почечные, сердечно-сосудистые и цереброваскулярные осложнения могут привести к инвалидизации, смерти.
Диагностика
При подозрении на болезнь Фабри обследование пациента проводится врачами нескольких специальностей: неврологом, кардиологом, дерматологом, генетиком. Диагноз устанавливают на основе типичных клинических проявлений, отягощенного семейного анамнеза, а также по данным лабораторной и инструментальной диагностики. Болезнь Фабри важно дифференцировать с коллагенозами, фибромиалгиями, болезнью Рейно. Специфические методы исследования включают:
- Анализ активности альфа-галактозидазы. Исследованию подвергается кровь (лейкоциты), сухие пятна крови, материал биопсии почек, культура кожных фибробластов. У мужчин активность фермента снижена. У женщин могут определяться показатели, соответствующие норме, нижней границе нормы или незначительно сниженные.
- Количественный анализ сфинголипидов. Выполняется исследование количества глоботриазилсфингозина в плазме крови, сухих пятнах крови. Тест широко применяется при скрининговых обследованиях. Высокие показатели с большой вероятностью указывают на наличие заболевания. Результаты используются не только для подтверждения диагноза, но и для контроля эффективности лечения.
- Секвенирование ДНК. У больных мужчин мутации диагностируются в гемизиготном наборе, у болеющих женщин или женщин-носителей – в гетерозиготном. Исследуется ген GLA. Обнаружение в его структуре мутационных изменений является самым точным способом диагностики болезни Фабри, особенно в отношении женщин.
- МРТ головного мозга. На снимках Т2 часто присутствует высокоинтенсивный сигнал в белом веществе теменной и фронтальной коры. На Т1-взвешенном изображении выявляется гиперинтенсивный сигнал в сером веществе глубоких структур. Болезнь характеризуется изолированным поражением заднего бугорка таламуса. Кроме того, выявляются сосудистые мальформации.
- ЭКГ, Эхо-КГ, МРТ сердца. Типична гипертрофия левого желудочка, на раннем этапе заболевания – укорочение интервала P-R, на позднем этапе – предсердно-желудочковая блокада. На МРТ визуализируется позднее попадание контраста на внутреннюю поверхность левого желудочка.
Лечение болезни Фабри
Единственным эффективным методом борьбы с заболеванием является ферментозаместительная терапия. С начала 2000-х годов лечение проводится рекомбинантными препаратами α-галактозы A. Своевременная медикаментозная терапия позволяет снизить выраженность невропатических болей, уменьшить выраженность гипертрофии левого сердечного желудочка, восстановить функциональность почек. Для скорейшего улучшения самочувствия больных применяются симптоматические средства. Парестезии и боли купируются антиконвульсантами, местными средствами с лидокаином.
При недостаточности функций почек и артериальной гипертонии назначаются ингибиторы АПФ, блокаторы АТ1-рецепторов, гемодиализ. Для профилактики тромбозов и инсультов используются антиагреганты, при тахикардии – антиаритмические препараты. Методы лечения продолжают разрабатываться. В настоящее время потенциально успешным считается направление генной инженерии. Предполагается, что вскоре станет возможным внедрение в клетки человеческого организма структурно правильного гена, задающего производство альфа субъединицы галактозидазы A.
Прогноз и профилактика
Своевременное начало терапии обеспечивает благоприятное течение болезни: симптомы купируются полностью либо остаются слабовыраженными, качество жизни пациентов повышается, женщины становятся способными зачать и выносить ребенка. Без лечения средняя продолжительность жизни мужчин составляет 40-60 лет, женщин – 40-70 лет. Профилактические меры заключаются в выявлении носительства мутации, медико-генетическом консультировании пар, в которых имеется партнер с подтвержденным диагнозом или отягощенным семейным анамнезом. При наступлении беременности таким парам необходимо проведение преимплантационной и пренатальной диагностики.
Что такое болезнь Фабри? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, гинеколога-эндокринолога со стажем в 11 лет.

Определение болезни. Причины заболевания
Болезнь Фабри (Андерсона-Фабри) — это редкое генетически детерминированное заболевание, которое характеризуется снижением фермента альфа-галактозидазы А, который отвечает за разрушение сфинголипида глоботриаозилцерамида, накопление которого в организме приводит к изменению функционирования клеток, прогрессирующему повреждению организма, что сопровождается разнообразными клиническими симптомами. Входит в группу лизосомальных болезней накопления, сфинголипидозов. [3]
Распространенность болезни Фабри составляет от 1 на 40000 до 1 на 120000 новорожденных. [4] Разница в данных объясняется трудностями постановки диагноза и разной частотой в популяциях.
В 1898 году дерматовенеролог Джон Фабри (J. Fabri) впервые описал случай узелковой пурпуры, впоследствии осложненной макроальбуминурией у тринадцатилетнего мальчика.
Заболевание обусловлено наличием мутации в Х-хромосоме. Мутация находится в гене GLA, локализованном в Хq22. [3]
Симптомы болезни Фабри
Основные диагностические критерии:
- жгучие боли в дистальных отделах конечностей;
- характерные кожные проявления;
- катаракта;
- кератопатия;
- накопление тригексозилцерамида в различных тканях;
- снижение активности фермента альфа-галактозидазы А в плазме крови, лейкоцитах, слезной жидкости и культуре фибробластов. [1]
Первые признаки заболевания, проявляющиеся постоянными нейропатическими болями, резкие эпизоды акропарестезий, непереносимость высоких и низких температур и чувство вялости проявляются еще в раннем школьном возрасте. [5]
Больные мужчины характеризуются специфической внешностью: выступающие супраорбитальные дуги и лобные бугры, прогнатия, пухлые губы и запавшая переносица. [2]
Подробная характеристика клинических симптомов
Типичными для данного заболевания являются мучительные приступы акропарестезии, при которых больной ощущает чувство жжения, покалывания, дискомфорта в дистальных отделах конечностей, возникающие при небольшом болевом раздражении. Тяжесть таких приступов с годами значительно возрастает, приступы случаются чаще и становятся продолжительнее. Иногда приступы такой боли длятся по несколько дней, сопровождаются субфебриллитетом, признаками воспаления по анализу крови. [4] Для болезни Андерсона-Фабри характерно наличие болевых кризов — возникающих периодически интенсивных болевых ощущений, которые воспринимаются как жжение, покалывание, онемение, локализуются в дистальных отделах конечностей, часто распространяются на проксимальные отделы конечностей, туловище. Спровоцировать ухудшение состояния могут стрессовые состояния, перегрев, переохлаждение, изменения атмосферного давления, физическая нагрузка, усталость. [2]
Ангиокератома является типичным поражением сосудов при болезни Фабри. Состоит из конгломерата нескольких расширенных сосудов, покрытых поверхностными слоями кожи. Локализуется на пальцах верхних и нижних конечностей, области вокруг губ, в районе гениталий, ануса, области вокруг пупка, на коленях. Не изменяет цвета при давлении. Представляет из себя пятна вишневого цвета, слегка приподнятые над поверхностью кожи, безболезненные. С годами возрастает их количество и размер. Ангиокератома обычно манифестирует в раннем возрасте и прогрессирует со временем.
Возможно развитие ангиэктазий на слизистых оболочках полости рта и глаз. [2]
К офтальмологическим симптомам относят увеличение диаметра и извилистый ход сосудов сетчатки и конъюнктивы, помутнение роговицы. При осмотре больных при помощи щелевой лампы иногда визуализируются светлые вихреподобные отложения субстрата в роговице, развивается воронкообразная кератопатия. В начальных стадиях заболевания острота зрения остается неизменной, но со временем развивается значительное помутнение роговицы, что может приводить к слепоте. Поражение зрительного нерва происходит редко, но несет за собой тяжелые последствия.
У пациентов с болезнью Андерсона-Фабри наблюдается нарушение потоотделения по типу гипогидроза (пониженного потоотделения), а иногда и ангидроза (отсутствия потоотделения). Это может приводить к непереносимости высоких температур, явлению перегрева организма.
Многие больные, особенно мужчины, жалуются на плохую переносимость физических нагрузок, утомляемость, что связано с быстрым перегревом, усилением интенсивности парестезий. [1]
С возрастом в патологический процесс вовлекается сердечно-сосудистая система. Депонирование сфинголипидов в кардиомиоцитах и эндотелии сосудов нарушает их функции, приводит к развитию фиброза. Проявляется это чувством одышки, стенокардитическими болями, аритмиями, тахикардией, гипертрофией левого желудочка, выявляемой по УЗИ, МРТ и ЭКГ. Это является наиболее неблагоприятным в прогностическом плане проявлением болезни.
Поражение почек выявляется у всех гемизигот. Прогрессирует ХПН, которая на начальных стадиях имеет скрытое течение, нет повышения артериального давления, выявляется нормальный или слегка повышенный уровень креатинина в сыворотке крови, небольшая протеинурия. Со временем почечная недостаточность достигает терминальной стадии, возникает тяжелая уремия, артериальная гипертензия.
Одними из самых явных проявлений заболевания являются неврологические нарушения. Больные предъявляют жалобы на приступы головокружения, головные боли, потерю сознания. Из-за депонирования сфинголипидов в лизосомах нервных клеток и эндотелии сосудов возникают ишемические состояния головного мозга, в результате чего у больных случаются транзиторные ишемические атаки, ишемические и геморрагические инсульты. Зачастую ишемический инсульт в молодом возрасте является единственным проявлением заболевания, особенно у женщин-гетерозигот и больных с атипичным типом заболевания. [3]
В результате ишемических изменений головного мозга больные становятся забывчивыми, рассеянными, неряшливыми, страдают когнитивные функции.
Больные могут жаловаться на шум, звон в ушах, резкое снижение остроты слуха, доходящей до глухоты. Наблюдается нейросенсорная тугоухость. [4]
Поражение желудочно-кишечного тракта проявляется болью в области живота, диспепсией, поносами, метеоризмом, запорами. У большинства больных эпизоды диареи сменяются запорами, развивается геморрой. Иногда развиваются желудочно-кишечные кровотечения. [1]
У части пациентов наблюдается легкая железодефицитная анемия, не требующая коррекции. [1]
У мужчин отмечается отставание физического и полового развития.
Зачастую пациентам с болезнью Андерсона-Фабри ставят диагноз "ревматическая лихорадка", что связано с эпизодами субфебриллитета и повреждением костной системы, которое проявляется вовлечением в патологический процесс дистальных межфаланговых суставов, снижением минеральной плотности позвонков и асептическими некрозами головок бедренной и таранной костей.
При тяжёлом течении заболевания, мучительных парестезиях нарушается психо-эмоциональная сфера человека, что приводит к депрессивным, тревожным состояниям. В некоторых случаях при интенсивных болевых проявлениях болезни пациенты имеют склонность к суициду.
Патогенез болезни Фабри
Болезнь Фабри наследуется по Х-сцепленному рецессивному типу наследования с полной пенетрантностью и различной экспрессивностью у мужчин (гемизигот). [1] Хотя некоторые авторы предполагают Х-сцепленный доминантный тип с неполной пенетрантностью. Тип наследования в настоящее время является темой для дискуссии у специалистов. [2]
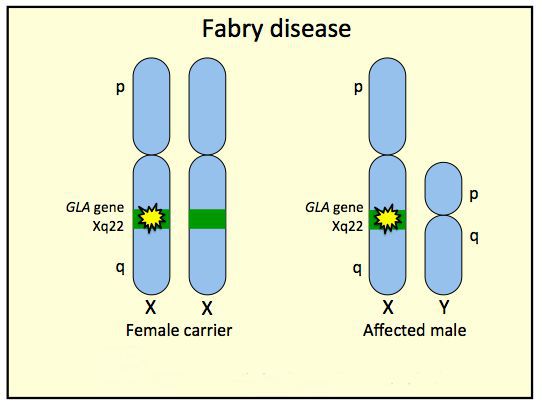
У мужчин-гемизигот заболевание протекает с более выраженными симптомами, как и у женщин-гомозигот. У женщин-гетерозигот зачастую обнаруживается атипичная форма болезни, либо они оказываются здоровыми. [3]
Заболевание вызывается мутацией в гене GLA, локализованном в коротком плече Х-хромосомы (Хq22). Этот ген кодирует синтез фермента альфа-галактозидазы А, ответственного за расщепление сфинголипида глоботриаозилцерамида. Глоботриаозилцерамид в своей структуре несет терминальный остаток альфа-галактозила, для отщепления которого требуется фермент альфа-галактозидаза А. При дефекте гена, кодирующего этот фермент, происходит нарушение этого процесса, что сопровождается отложением гликосфинголипидов с терминальным α-галактозил остатком в клетках.
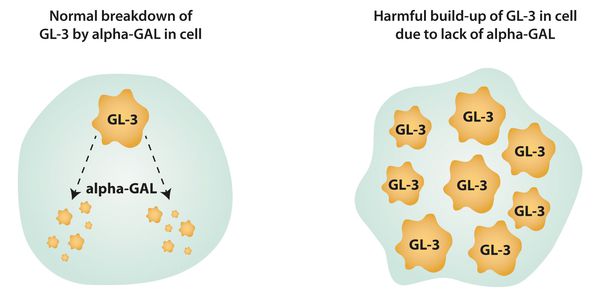
Сфинголипиды — это липиды, производные алифатических аминоспиртов. В случае, когда их разрушение нарушается, происходит избыточное накопление церамидтригексозида в лизосомах клеток.
Накопление субстрата в клетках значительно нарушает их жизнедеятельность, что приводит к изменению функций и клиническим проявлениям заболевания, нарушается нервная передача, связь между клетками.
В сосудистой стенке, почках, миокарде накапливаются сфинголипиды, индуцирующие в последующем процессы фиброгенеза, итогом которых является органная недостаточность. [2]
При накоплении субстрата в эндотелиальной ткани сосудов происходит их сужение, наблюдается нарушение микроциркуляции, гипоксия тканей. Это основной механизм развития ишемических нарушений.
Накопление сфинголипида в нервных клетках приводит к их структурным нарушениям, проявляющимся увеличением числа кальциевых каналов и образованием патологических ноцицептивных связей, повышению возбудимости путей болевой чувствительности, и развитию типичной симптоматики. [2]
Развитие гипогидроза, как и ангидроза, связано с депонированем сфинголипидов в клетках потовых желез, нарушенной иннервацией и уменьшенным кровоснабжением кожи. [2]
Накопление тригексозилцерамида в миокарде вызывает развитие фиброза, [8] прогрессирующей гипертрофической кардиомиопатии, а при наличии сужения коронарных сосудов, это приводит к ишемическим кардиальным симптомам, острому коронарному синдрому, развитию сердечной недостаточности.
Предполагается, что усиление боли при физических нагрузках связано со спазмом суженных сосудов, приводящему к ухудшению трофики нервных волокон. [2]
Отложение субстрата в роговице приводит к развитию ее помутнения, а поражение сосудов может способствовать ухудшению зрения, вплоть до слепоты.
Классификация и стадии развития болезни Фабри
Выделяют две основные формы болезни Фабри: [2]
- классическую;
- атипичную.
Классическая характеризуется ранней манифестацией заболевания (в первом десятилетии жизни), наличием характерных симптомов и осложнений.
При атипичной форме происходит изолированное поражение головного мозга (ранние инсульты), сердца и почек, она манифестирует в более позднем возрасте и представляет большие трудности для диагностики.
Осложнения болезни Фабри
Со стороны нервной системы:
- ишемические инсульты в молодом возрасте, нередко являющиеся причиной смерти.
Со стороны сердечно-сосудистой системы:
- ишемические инфаркты;
- кардиомиопатии;
- аритмии;
- сердечная недостаточность, приводящая к смерти.
Со стороны почек:
- терминальная стадия почечной недостаточности, требующая трансплантации почек.
Другие системы:
- нарушение зрения; слуха, вплоть до глухоты;
- переломы костей;
- перегрев организма.
Смерть чаще всего наступает от уремии или ишемических поражений мозга и сердца на четвертом десятилетии жизни. [8]
Диагностика болезни Фабри
Ранняя диагностика очень важна для правильного и своевременного назначения лечения пораженных болезнью органов, предотвращения осложнений. [5]
Важным методом диагностики болезни Фабри является оценка генеалогического анамнеза пациента, так как при этом могут обнаружиться родственники со сходными симптомами, которые не знают о своем заболевании. Ген, ответственный за болезнь Андерсона-Фабри, может передаваться через большое число поколений, поэтому больными оказываются многие ближние и дальние родственники. Для определения риска наследования этого заболевания необходимо собрать информацию о здоровье всех известных членов семьи. [3]
Болезнь Фабри бывает очень трудно отличить от более распространенных заболеваний, и пациенты в течение долгих лет могут оставаться без верного диагноза.
Предварительный диагноз ставится по результатам опроса, жалоб пациента, генеалогическому анамнезу.
Для верификации диагноза используют методы обнаружения субстратов и энзимов, ДНК-диагностику.
- Измерение активности альфа-галактозидазы А.[2] При болезни Андерсона-Фабри активность этого фермента у представителей мужского пола всегда ниже нормы, а у женщин этот показатель бывает в пределах нормы или слегка снижен. Материалом для проведения исследования могут служить лейкоциты, плазма крови, слезная жидкость, культура фибробластов.
- Наиболее точным методом диагностики является секвенирование гена GLA. [2] На сегодняшний день описано более пятисот мутаций этого гена, приводящих к болезни Фабри. Использование данной методики ограничено её стоимостью. ДНК-диагностику целесообразно применять для женщин, т. к. у них определение активности альфа-галактозидазы А не всегда дает достоверный результат, и для родственников больного, т. к. они могут являться носителями мутантного гена или больны атипичной формой заболевания. Проведение данного анализа возможно в Центре молекулярной генетики в Москве.
- Количественное определение глоботриаозилцерамида. Этот метод зарекомендовал себя для наблюдения за состоянием пациентов и оценки эффективности лечения, для определения формы заболевания (типичная, атипичная). Материалом для исследования могут быть плазма крови или сухие пятна крови.
- В некоторых случаях проводится биопсия почки с целью обнаружения клеток, содержащих лизосомы с характерным субстратом.
Дифференциальная диагностика болезни Фабри проводится с наследственной геморрагической телеангиэктазией, ревматической лихорадкой, болезнью Фордайса, Шиндлера, другими наследственными болезнями накопления. [1]
Лечение болезни Фабри
Лечение болезни Фабри состоит в замещении дефицитного фермента с помощью ферментозаместительной терапии. Она проводится с помощью внутривенного вливания препарата. Обычно ферментозаместительная терапия используется вместе с различными методами лечения конкретных симптомов. [5]
В России сегодня используются два препарата для ферментозаместительной терапии: Фабразим, Джензайм; Реплагал, Шайер. Эти препараты имеют очень большую стоимость, лечение больных оплачивается из средств федерального бюджета. Так как заболевание относится к орфанным (редким), существует закон об ускоренной процедуре исследования лекарственных препаратов, предназначенных для лечения таких болезней. [3]
Мужчинам начало ферментозаместительного лечения рекомендуется в ближайшее время после установления диагноза. [5]
Для купирования боли применяются препараты из групп атиконвульсантов, антидепрессантов, анальгетиков, нестероидных противовоспалительных препаратов, наркотических анальгетиков. [2]
Больным показано применение антигипертензивных препаратов.
При развитии терминальной почечной недостаточности проводят трансплантацию почки. Донорами не рекомендуется быть женщинам, которые являются родственницами больного. [2]
В качестве коррекции тугоухости показано использование слуховых аппаратов.
Физическая активность пациентов с болезнью Фабри должна быть ограничена из-за возможного усиления симптомов и перегрева организма. [3]
Ситуации, при которых нецелесообразно назначать ферментозаместительную терапию:
- период беременности и лактации;
- если имеется другое опасное для жизни заболевание, при котором прогноз от применения заместительной терапии не станет лучше;
- есть серьезные осложнения (ишемический инсульт, реанимационные больные).
Прогноз. Профилактика
При надлежащем лечении и вовремя начатой заместительной ферментативной терапии прогноз для больных благоприятный. При отсутствии лечения смерть от сердечно-сосудистых, неврологических и почечных осложнений наступает на четвертом десятилетии жизни. [4]
Профилактика заболевания заключается в пренатальной или предимплантационной (при ЭКО) диагностике наличия мутантного гена методом ДНК-диагностики и прерывания беременности по медицинским показаниям. [4] Необходимо обследование всех потенциальных носителей гена для своевременного назначения заместительной ферментативной терапии и предотвращения клинических проявлений болезни и ее осложнений. Возможно скрининговое исследование сухих пятен крови с определением активности альфа-галактозидазы, но оно ограничено финансовыми возможностями региона. Необходимо, чтобы все пациенты с признаками болезни Фабри были исследованы на это заболевание. Для этого существует программа, регламентирующая забор и отправку материала в Центр молекулярной генетики, обеспечение больных препаратами заместительной терапии.
Читайте также: