
Паркинсонизм при нормотензивной гидроцефалии
Паркинсонизм – синдром, связанный с поражением базальных ганглиев и проявляющийся гипокинезией и ригидностью, которые часто сопровождаются тремором покоя и постуральными нарушениями [1, 8]. Основной нозологической формой паркинсонизма является болезнь Паркинсона (БП), на долю которой приходится около 70 % его случаев [1, 6, 9]. БП в ряду других нозологических форм паркинсонизма отличается более длительным доброкачественным течением и более высокой эффективностью противопаркинсонических, в первую очередь дофаминергических средств. Лечение БП – сложный многоэтапный процесс, имеющий целью многолетнее поддержание оптимального уровня мобильности пациентов. Проблемы лечения БП подробно освещались в публикациях последних лет [1, 7, 18].
Другие формы паркинсонизма характеризуются более быстрым прогрессированием и часто приводят к утрате способности к самостоятельному передвижению в течение первых 5 лет заболевания. Симптомы паркинсонизма при этих заболеваниях в значительно меньшей степени реагируют на противопаркинсонические средства или не реагируют на них совсем [3, 17]. Проблемы их лечения обсуждаются редко, и это зачастую приводит к тому, что существующие, пусть и весьма ограниченные возможности помощи таким пациентам не используются в достаточной мере, что приводит к более быстрой утрате мобильности и повышению вероятности раннего летального исхода. В статье рассмотрены некоторые практические вопросы диагностики и лечения паркинсонизма, возникающего в рамках иных, чем БП, заболеваний.
Нозологическая структура паркинсонизма
Алгоритм нозологической диагностики паркинсонизма
Лекарственный паркинсонизм
Лекарственный паркинсонизм может быть вызван нейролептиками или другими препаратами, блокирующими дофаминовые D2-рецепторы (например, циннаризином, метоклопрамидом) либо симпатолитиками (например, резерпином), истощающими пресинаптические запасы дофамина. В пользу лекарственного паркинсонизма свидетельствуют:
• анамнестические данные о длительном приёме препарата, способного вызвать паркинсонизм;
• подострое развитие и быстрый темп прогрессирования паркинсонизма;
• двустороннее начало и относительная симметричность симптоматики;
• отсутствие типичного тремора покоя (чаще наблюдается постуральное дрожание, иногда, правда, сохраняющееся и в покое);
• комбинация с другими дискинезиями, например, акатизией, стереотипиями, орофациальными дискинезиями [5].
Главный признак лекарственного паркинсонизма – возможность регресса симптомов в течение нескольких недель после отмены соответствующего препарата. Но иногда регресс симптомов занимает месяцы и даже годы, особенно после применения препаратов длительного действия. Следует учитывать, что скрыто протекающее нейродегенеративное заболевание (от БП до гепатолентикулярной дегенерации) повышает риск экстрапирамидных осложнений. В этих случаях симптомы паркинсонизма даже после отмены нейролептика продолжают неуклонно нарастать (иногда после непродолжительного улучшения).
При появлении лекарственного паркинсонизма следует отменить вызвавший его препарат или заменить его другим средством, не блокирующим дофаминовые рецепторы в головном мозге (например, стандартный нейролептик заменить атипичным, а метоклопрамид – домперидоном). Для коррекции двигательных нарушений можно назначать любой противопаркинсонический препарат, но, учитывая возможность ухудшения первичного заболевания (особенно психиатрического), препараты леводопы и агонисты дофаминовых рецепторов не назначают, а используют, главным образом, холинолитики и амантадин. Учитывая нередкие побочные эффекты холинолитиков, в т. ч. возможность стойкого ухудшения когнитивных функций, амантадин в последние годы становится препаратом выбора в лечении лекарственного паркинсонизма, особенно у пожилых. Препараты леводопы иногда назначают на непродолжительное время, если больной не страдает психиатрическим заболеванием и принимал нейролептик как противорвотное средство, а его отмена не привела к быстрому регрессу симптомов. В качестве вспомогательных средств используют витамины В6 и Е [8].
Сосудистый паркинсонизм
В прошлом сосудистое поражение мозга считалось основной причиной паркинсонизма, однако патоморфологические исследования показывают, что оно является причиной лишь 3–6 % случаев паркинсонизма. Диагностика сосудистого паркинсонизма (СП) значительно облегчается с помощью методов нейровизуализации, однако выявление у больного паркинсонизмом при КТ и МРТ сосудистого поражения мозга является необходимым, но не достаточным условием диагностики СП – причинно-следственную связь между цереброваскулярным заболеванием и паркинсонизмом нужно доказывать. В пользу такой связи свидетельствуют не столько сосудистые факторы риска, клинические признаки цереброваскулярного заболевания (инсультов или дисциркуляторной энцефалопатии), сколько:
1) особенности паркинсонического синдрома: сравнительно малая эффективность препаратов леводопы, преимущественное вовлечение нижних конечностей, отсутствие классического тремора покоя и позы просителя, раннее развитие постуральной неустойчивости, деменции, псевдобульбарного синдрома, наличие мозжечковой или пирамидной симптоматики и других симптомов не характерных для БП;
2) особенности течения заболевания – острое или подострое развитие вскоре после перенесённого инсульта с последующей стабилизацией и регрессом симптомов либо ступенеобразное прогрессирование с чередованием эпизодов быстрого нарастания симптомов и их последующего частичного регресса;
3) выявление при КТ или МРТ структурных изменений головного мозга, способных вызвать синдром паркинсонизма, т. е. расположенных в стратегических для развития паркинсонизма зонах (обширный подкорковый лейкоареоз, множественные двусторонние подкорковые очаги, поражения среднего мозга, таламуса или лобных долей) [2].
Нормотензивная гидроцефалия
Нормотензивная гидроцефалия (НТГ) – разновидность сообщающейся гидроцефалии, при которой расширение желудочков происходит в отсутствие клинических признаков внутричерепной гипертензии. НТГ может быть следствием нарушения всасывания ликвора арахноидальными ворсинами над конвекситальной поверхностью полушарий головного мозга, возникающего в исходе субарахноидального кровоизлияния, гнойного менингита, тяжёлой черепно-мозговой травмы или в силу других причин. Как и при дисциркуляторной энцефалопатии, при НТГ чаще встречается не истинный акинетико-ригидный синдром, а нарушения ходьбы по типу лобной дисбазии. Отличительными особенностями паркинсонизма при НТГ являются быстрое прогрессирование, сопутствующая деменция подкорково-лобного типа, нарушения мочеиспускания, паралич взора вверх. При МРТ выявляются расширение желудочковой системы, диспропорциональное расширению корковых борозд, наличие признаков усиленного тока цереброспинальной жидкости через третий желудочек и сильвиев водопровод (по данным МРТ).
Реакция на препараты леводопы чаще отсутствует или имеет преходящий характер. Но некоторые случаи паркинсонизма при гидроцефалии напоминают болезнь Паркинсона – по наличию тремора покоя и хорошей реакции на препараты леводопы, а также по развитию при длительном приёме леводопы лекарственных дискинезий и моторных флуктуаций. В подобных случаях паркинсонизм, по-видимому, обусловлен давлением, оказываемым расширенными желудочками на нигростриарные пути [8]. Важное диагностическое значение имеет положительный результат ликвородинамической пробы: удаление 40–50 мл цереброспинальной жидкости при люмбальной пункции может приводить к существенному улучшению двигательных функций в течение нескольких часов или дней.
Лечение нейрохирургическое; оно заключается в устранении препятствия для оттока ликвора или установлении вентрикуло-перитонеального (вентрикуло-атриального или вентрикуло-венозного) шунта. В ряде случаев признаки паркинсонизма уменьшаются под влиянием препаратов леводопы, что указывает на преимущественное повреждение нигростриарных путей. Часто требуется лишь кратковременное применение препаратов леводопы до получения клинического эффекта от шунтирования или устранения окклюзии, но иногда потребность в леводопе сохраняется в течение многих лет после операции.
Прогрессирующий надъядерный паралич
Мультисистемная атрофия
Мультисистемная атрофия (МСА) – спорадическое заболевание, которое характеризуется преимущественным вовлечением базальных ганглиев, стволово-мозжечковых систем, вегетативных нейронов ствола и спинного мозга и клинически проявляется сочетанием паркинсонизма с вегетативной недостаточностью, мозжечковым и пирамидным синдромами. В основе лежит накопление альфа-синуклеина в олигодендроцитах [1, 8, 9].
Заболевание чаще всего дебютирует на шестом десятилетии жизни. Выделяют три основных клинических типа МСА. При преобладании паркинсонизма диагностируют стриатонигральный (паркинсонический) тип, при преобладании мозжечковой атаксии – оливопонтоцеребеллярный (мозжечковый) тип, при преобладании признаков быстро нарастающей вегетативной недостаточности, прежде всего, ортостатической гипотензии – синдром Шая–Драйджера (вегетативный тип). Паркинсонизм возникает у 90 % больных МСА и в целом напоминает синдром, наблюдающийся при БП, однако он отличается малоэффективностью леводопы, быстрым прогрессированием с относительно ранним развитием постуральной неустойчивости, нарушений позы, псевдобульбарных нарушений. Как и при БП, паркинсонические симптомы в большинстве случаев имеют асимметричный характер. У большинства больных отмечается тремор, но классический тремор покоя наблюдается только в 10 % случаев. Часто выявляется иррегулярный постурально-кинетический тремор, возникающий вследствие наложения на дрожательный гиперкинез лёгких миоклонических подергиваний пальцев (миоклонический тремор) [12, 19].
Для МСА характерно более раннее развитие изменений позы, чем для БП, особенно характерен (хотя и не специфичен) антероколлис, реже встречаются другие позные изменения, например, по типу камптокормии или синдрома Пизанской башни. Вегетативная недостаточность выявляется у всех больных (в её отсутствие диагноз МСА теряет достоверность). Помимо ортостатической гипотензии, она может проявляться недержанием или задержкой мочи, ослаблением моторики желудочно-кишечного тракта, акрогипотермией [20]. Относительно специфичны для МСА затруднения дыхания, одной из причин которых является дисфункция (слабость или дистония) мышц, отводящих голосовые складки, которая может проявляться инспираторным стридором, особенно выраженным в ночное время. Хотя у многих пациентов отмечаются депрессия и другие аффективные нарушения, интеллект у большинства пациентов остается относительно сохранным, однако на поздней стадии заболевания может развиваться деменция. При МРТ в Т2-режиме выявляется характерное снижение интенсивности сигнала от скорлупы, иногда вместе со щелевидными полосками гиперинтенсивности по её наружному краю. Кроме того, нередко наблюдаются атрофия мозжечка и моста, изменение интенсивности сигнала от понтоцеребеллярных волокон [3].
Хотя в большинстве случаев леводопа неэффективна, не менее чем у трети больных на фоне лечения отмечается умеренное уменьшение симптомов, которое чаще всего, в отличие от БП, бывает нестойким. Лишь у 10 % больных (по-видимому, с опережающей дегенерацией нигростриарных нейронов) эффективность препаратов леводопы сохраняется в течение всего заболевания. У некоторых больных симптомы паркинсонизма уменьшаются лишь при применении высоких доз леводопы, а их эффект иногда может проявиться лишь спустя несколько недель. Поэтому делать заключение о неэффективности препаратов леводопы при МСА следует лишь после 2–3 месяцев лечения, при этом доза леводопы должна быть доведена как минимум до 1000–1200 мг/сут (например, 5–6 таблеток мадопара 200/50). В части случаев больные МСА (особенно при синдроме Шая–Дрейджера) не способны переносить даже низкие дозы леводопы из-за усиления ортостатической гипотензии. В этих случаях коррекция ортостатической гипотензии позволяет достичь эффективной дозы. У большинства больных леводопа не вызывает дискинезии, если же они возникают, то имеют преимущественно дистонический характер и вовлекают аксиальную мускулатуру (включая лицо и шею), но не конечности, как при БП.
Спутанность сознания, галлюцинации и параноидный синдром возникают значительно реже, чем при БП. В отсутствие эффекта дозу леводопы следует снизить, но полная отмена препарата нередко приводит к ухудшению состояния даже при кажущемся отсутствии его эффекта. Повторное назначение леводопы в этих случаях нередко оказывается безуспешным.
Агонисты дофаминовых рецепторов также эффективны примерно у трети больных, особенно на ранней стадии заболевания. Однако если препарат леводопы в максимальной дозе оказался неэффективным, то добавление агониста дофаминовых рецепторов редко приносит дополнительный эффект. Амантадин в дозе 200–500 мг/сут улучшает состояние у 10–20 % больных, чаще на ранней стадии заболевания. Некоторым больным он помогает больше, чем леводопа. У части больных умеренное действие оказывают холинолитики, но они противопоказаны при затрудненном мочеиспускании и накоплении остаточной мочи в мочевом пузыре. При треморе и миоклонии дополнительно назначают клоназепам (0,5–1,0 мг 2 раза в день) Показаны регулярные занятия ЛФК, направленные на поддержание мобильности больного, улучшение ходьбы, обучение приёмам предотвращения падений. При антероколлисе применяют поддерживающий воротник; иногда больные лучше переносят вынужденное положение головы, если надевают очки со специальными призматическими стеклами, позволяющими им смотреть перед собой. Инъекции ботулотоксина в передние группы мышц шеи при антероколлисе обычно не проводят из-за высокого риска дисфагии.
Важное значение имеет коррекция вегетативной недостаточности, особенно ортостатической гипотензии, которая иногда в большей степени ограничивает активность больного, чем собственно двигательные расстройства. В первую очередь осуществляют немедикаментозные меры: увеличение потребления соли и жидкости (в отсутствие сердечной недостаточности), ношение эластичных чулок или эластичное бинтование (до верхней трети бедра, иногда до живота), поднятие изголовья постели во время ночного сна на 15–20 см. Нужно избегать провоцирующих факторов: натуживания (при склонности к запорам рекомендуют продукты с высоким содержанием пищевых волокон, слабительные), резких изменений положения тела, тепловых процедур и перегревания, приёма алкоголя, длительного постельного режима, интенсивных физических упражнений, особенно в изометрическом режиме. Следует по возможности отменить гипотензивные и сосудорасширяющие средства, симпатолитики, диуретики, трициклические антидепрессанты или уменьшить их дозу.
Если перечисленные меры оказались недостаточно эффективными, прибегают к медикаментозным средствам, повышающим объём циркулирующей крови и сосудистый тонус. Наиболее эффективное из них – фторсодержащий синтетический кортикостероид флудрокортизон (кортинефф), обладающий минералокортикоидной активностью. Лечение начинают с 1 таблетки (0,1 мг) утром, затем дозу постепенно повышают каждые 2 нед. до достижения эффекта (в среднем до 0,2–0,4 мг/сут в 2 приёма). Для уменьшения ортостатической гипотензии, обычно в комплексе с флудрокортизоном, назначают и симпатомиметики, в первую очередь агонист a1-адренорецепторов мидодрин, начиная с 2,5 мг 2 раза в сут до 15–30 мг/сут в 2–3 приёма (в первой половине дня) [20].
При императивных позывах у больных с гиперрефлекторным мочевым пузырем эффективны препараты с холинолитическим действием: оксибутинин, толтерадин, троспий. Поскольку эти препараты могут вызвать затруднения мочеиспускания, лечение следует проводить под контролем остаточной мочи. При нарушении опорожнения мочевого пузыря и накоплении остаточной мочи по возможности следует отменить холинолитики и назначить селективные блокаторы 1-адренорецепторов, расслабляющие внутренний сфинктер (например, тамсулозин или доксазозин). При назначении этих препаратов следует соблюдать особую осторожность, так как они могут усилить ортостатическую гипотензию.
При депрессии показано назначение антидепрессантов (амитриптилин, пароксетин, сертралин и др.). Показано, что некоторые антидепрессанты (например, пароксетин) могут улучшать двигательные функции при МСА. При психомоторном возбуждении во сне с быстрым движением глаз показаны небольшие дозы клоназепама (0,5–2 мг/сут). При дыхательном стридоре, который может быть фактором внезапной смерти, могут быть эффективны аппараты, создающие постоянное положительное давление в дыхательных путях. В резистентных случаях показана трахеостомия, но лишь на ранней стадии МСА, когда остальные неврологические функции более сохранны.
Деменция с тельцами Леви
Нормотензивная гидроцефалия является формой сообщающейся гидроцефалии. Приблизительно одна треть больных с указанным расстройством имеет в анамнезе спонтанное или травматическое субарахноидальное кровоизлияние, менингит, которые могли привести к нарушению механизмов абсорбции ЦСЖ над поверхностью мозга. Хотя давление ЦСЖ, измеренное при люмбальной пункции, в норме, существует избыточное давление на стенки расширенных боковых желудочков, особенно в передних рогах, ведущее к компрессии окружающих структур.
Применение методов нейровизуализации при нормотензивной гидроцефалии. Наблюдается расширение боковых желудочков (особенно передних и боковых рогов), которое происходит непропорционально выраженности корковой атрофии. МРТ обнаруживает перивентрикулярную гиперинтенсивность протонной плотности, свидетельствующую о наличии трансэпендимального тока ЦСЖ. Разграничение данных признаков с неспецифическим увеличением перивентрикулярных изменений в Т2-режиме у пожилых людей может быть затруднительным.

Изотопная цистернография, хотя и демонстрирует нарушение абсорбции ЦСЖ у некоторых пациентов, не рассматривается как надежный метод прогнозирования эффекта шунтирования. с Другие тесты. Тест Фишера состоит в удалении 30-50 мл ЦСЖ и наблюдении за улучшением клинической картины в течение следующих 24 ч. Это информативный тест, который не требует какой-либо сложной лабораторной техники. Мониторирование внутричерепного давления позволяет наблюдать периоды высокого давления ЦСЖ (b-волны) и прогнозировать эффект шунтирования.
Гемиатрофия-гемипаркинсонизм. При этом заболевании сравнительно рано на стороне гемиатрофии появляются признаки асимметричного синдрома паркинсонизма. У таких больных в анамнезе имеются указания на патологические роды, а также признаки атрофии контралатерального полушария. В совокупности это увеличивает вероятность неврологических расстройств в раннем детстве, а в дальнейшем обнаруживаются явления паркинсонизма. Медленное прогрессировать заболевания, редкое сочетание с дистонией и выраженная асимметрия проявлений являются основой для разграничения с ИБП.
- МФТП (1 -метил-4-фенил-1,2,3,6-тетрагидропиридин). Неумышленный прием этого токсического вещества привел к развитию острого выраженного паркинсо-нического симптомокомплекса у нескольких лиц с наркотической зависимостью. В лабораториях МФТП широко применяется для моделирования ИБП у животных. Новые случаи паркинсонизма, обусловленного его приемом, у людей в настоящее время встречаются редко.
- Окись углерода (СО). Паркинсонизм может развиваться вследствие острого или хронического отравления СО. Это вещество вызывает некроз бледного шара и полосатого тела. Экстрапирамидные нарушения могут возникать внезапно сразу же после инцидента, однако значительно чаще развиваются в течение нескольких дней или недель после выхода пациента из комы. Анамнестические данные (интоксикация СО), недостаточный эффект леводопы или его отсутствие подтверждают диагноз.
- Интоксикация марганцем может приводить к развитию синдрома паркинсонизма. Она часто сопровождается появлением необычных поведенческих расстройств (галлюцинации и эмоциональная лабильность), а также других двигательных нарушений, например, дистонии.
- Интоксикация цианидами и метанолом также может вызывать двусторонние некрозы базальных ганглиев и явления паркинсонизма.
Цереброваскулярные заболевания. Лакунарное состояние с множеством мелких инфарктов в области базальных ганглиев, как и подострая атеросклеротическая энцефалопатия, сопровождаются нарушением связей базальных ганглиев, что приводит к развитию паркинсонизма. При этих состояниях также часто наблюдается деменция. Тремор покоя у таких больных обычно отсутствует. Нарушения ходьбы могут быть очень заметными и иногда являются единственным неврологическим расстройством, что дает основание для термина «паркинсонизм нижней части тела*. Применение леводопы дает ограниченный положительный эффект.
Травма. Травматическая энцефалопатия боксеров — это прогрессирующий неврологический симптомокомплекс, состоящий из паркинсонизма, деменции и атаксии. Он наблюдается у боксеров с повторными травмами головы в анамнезе. Лечение обычно малоэффективно. Острое очаговое повреждение среднего мозга и черной субстанции или субдуральная гематома являются двумя другими возможными причинами посттравматического паркинсонизма.
Энцефалит. Приблизительно 50 % выживших после летаргического эпидемического энцефалита в 1917—1925 годах в дальнейшем, часто через несколько десятилетий, заболевали паркинсонизмом. С тех пор данная разновидность энцефалита в эпидемической форме не наблюдается. Соответственно, новые случаи постэнцефалитического паркинсонизма в настоящее время редки.
*Импакт фактор за 2018 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Читайте в новом номере
ММА имени И.М. Сеченова
Клиническая картина

Болезнь Паркинсона. На рисунке А показан участок мозга здорового человека с хорошо пигментированным черным веществом. На рисунке В - участок мозга пациента, страдающего болезнью Паркинсона. Заметно отсутствие пигментации черного вещества.
П – это синдром, который может встречаться в рамках разных заболеваний. Наиболее часто причиной П является идиопатический паркинсонизм, или болезнь Паркинсона (БП). П часто наблюдается в рамках других идиопатических дегенеративных заболеваний нервной системы. Последние часто называют паркинсонизмом–плюс. К этой группе относятся мультисистемная атрофия, прогрессирующий надъядерный паралич, болезнь диффузных телец Леви, кортико-базальная дегенерация. Нередко встречается симптоматический П (не связанный с первичным дегенеративным заболеванием нервной системы). К нему относят лекарственный, сосудистый, посттравматический, постэнцефалитический, токсический П, П при опухолях головного мозга и гидроцефалии. П характеризует также клиническую картину некоторых наследственных дегенеративных заболеваний ЦНС. К их числу относятся гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова), спиноцеребеллярные атаксии, семейная кальцификация базальных ганглиев, болезнь Гентингтона и др. Таким образом, синдром П не является синонимом БП.
Диагноз и дифференциальный диагноз
Эссенциальный тремор
На первом этапе диагностической работы необходимо убедиться, что у больного действительно имеется синдром П. Наиболее часто за истинный П ошибочно принимают эссенциальный тремор (ЭТ) и своеобразные изменения походки, связанные с сосудистой патологией головного мозга. Таким пациентам очень часто неправильно ставят диагноз БП.
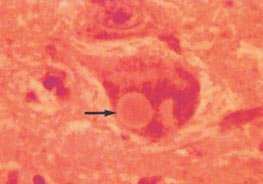
Тельца Леви при болезни Паркинсона. В цитоплазме нейрона определяется эозинофильное ядро окруженное неокрашенной зоной.
Последние, как правило, выявляют множественные мелкие очаги сосудистого происхождения (лакуны) в области базальных ганглиев и/или изменения белого вещества полушарий головного мозга (лейкоареоз). Следует иметь в виду, что схожая клиническая картина может возникать иногда при нормотензивной гидроцефалии и реже – при опухолевом поражении головного мозга. При нормотензивной гидроцефалии наряду с нарушением ходьбы по типу описанного выше имеют место деменция и расстройство функции тазовых органов (эту триаду симптомов называют триадой Хакима–Адамса). МРТ головы, как правило, обнаруживает резко расширенные боковые желудочки.
Болезнь Паркинсона
Диагноз БП – это клинический диагноз. Параклинические методы исследования обычно выявляют неспецифические изменения. Нередко у больных с классической БП в головном мозге имеются сосудистые изменения. Это, однако, ни в коем случае не является свидетельством сосудистого генеза болезни. Просто у большинства пожилых людей, в том числе практически здоровых, подобные изменения могут иметь место. Тем не менее, в некоторых случаях на БП наслаивается атеросклеротический псевдопаркинсонизм, что может быть причиной необычной для БП походки и ранних постуральных нарушений.
Лекарственный паркинсонизм
Лекарственный П может быть обусловлен препаратами, которые воздействуют на пресинаптические дофаминовые нейроны черной субстанции, истощая запасы дофамина в них (например, резерпин) или, наиболее часто, нейролептиками, которые блокируют постсинаптические дофаминовые рецепторы, такими как производные фенотиазина (хлорпромазин), бутирофеноны (галоперидол), тиоксантины (флупентиксол) и бензамиды (сульпирид). Эти препараты часто применяются при психических заболеваниях. Также П может быть вызван прохлорперазином (используется при рвоте, головокружении и неустойчивости), метоклопрамидом (применяется при заболеваниях желудочно-кишечного тракта, для купирования тошноты и рвоты). П может быть обусловлен также циннаризином, который является атипичным блокатором кальциевых каналов (применяется при вестибулярных расстройствах). Комбинация нейролептиков и антидепрессантов также может быть причиной П.
В психиатрических лечебницах лекарственный П встречается часто. Гипомимия и ахейрокинез – настолько обычное явление среди этого контингента больных, что психиатры часто даже не обращают на них внимание. Тремор встречается менее часто, но может иметь вид классического паркинсонического дрожания. Более того, лекарственный П может быть асимметричным, подобно БП, и нередко совершенно не отличим от БП. Одним из признаков, по которому следует заподозрить лекарственный П, является наличие наряду с акинетико-ригидным синдромом насильственных движений в виде, например, оро-мандибулярной дискинезии (непроизвольные жевательные и/или сосательные движения) или дистонических явлений (спастической кривошеи, окулогирных кризов), стереотипий, акатизии (неусидчивость). Нередко тяжелый лекарственный П сопровождается выраженной дизартрией и дисфагией. Индивидуальная чувствительность к дофаминблокирующим препаратам очень разнообразна. Некоторые пациенты без каких-либо проблем переносят длительное лечение большими дозами этих веществ, тогда как у других побочные явления развиваются уже на малых дозах. Чаще, однако, признаки лекарственного П возникают при приеме больших доз нейролептиков. Лекарственный П обычно развивается постепенно в течение дней или недель. У большинства больных первые признаки появляются через 3 нед после начала лечения. Наиболее часто встречающиеся дебютные признаки – гипомимия и недостаточное раскачивание рук во время ходьбы.
Течение лекарственного П может быть различным. В большинстве случаев он постепенно, в течение нескольких недель, а иногда и дней, проходит после прекращения приема вызвавшего его препарата. Тем не менее нередки случаи, когда П длится в течение месяцев, иногда почти год. Такая ситуация наблюдается при применении нейролептических препаратов, способных к депонированию. В редких случаях лекарственный П не проходит и продолжает прогрессировать, несмотря на прекращение приема вызвавшего его агента. Подобные случаи чаще встречаются среди пожилых людей. Считается, что в таких случаях прогрессирует не сам по себе лекарственный П, а начинает развиваться БП.
Мультисистемная атрофия
Мультисистемная атрофия (МСА) – это спорадическое заболевание, возникающее у взрослых лиц, при котором в отличие от БП дегенерации подвергается не только нигро-стриарная система, но также множество других образований ЦНС, включая мозжечок и его связи, пирамидные пути и образования вегетативной нервной системы (отсюда и происходит название болезни). Соответственно клинически МСА характеризуется сочетанием П, мозжечковых нарушений, пирамидных расстройств и прогрессирующей вегетативной недостаточности (ПВН). П при МСА обусловлен не только поражением клеток черной субстанции, что вызывает дефицит дофамина, но также дегенерацией тех постсинаптических рецепторов, с которыми должен взаимодействовать дофамин.
В клинической картине МСА может преобладать та или иная симптоматика. Те случаи, при которых на первый план выступает П, обозначают термином нигро-стриальная дегенерация (СНД); если в клинической картине ведущим является мозжечковый синдром, это состояние называют оливо-понто-церебеллярной атрофией (ОПЦА); случаи, когда ядром клинической картины является ПВН, обозначают эпонимическим названием – синдром Шая–Дрейджера (СШД).
Несмотря на все различия, нередки ситуации, когда МСА невозможно отличить от БП. В таких случаях следует в основном ориентироваться на эффект препаратов леводопы. При БП эти препараты оказывают драматический положительный эффект, тогда как при МСА этот эффект не столь выражен, кратковременен, а нередко отсутствует совсем. Это обусловлено поражением постсинаптических рецепторов, с которыми должна взаимодействовать леводопа.
Признаки, при наличии которых у больных с П следует подумать об МСА, следующие: быстрое прогрессирование симптоматики; рано развившиеся нарушения равновесия и падения; отсутствие улучшения при лечении препаратами леводопы или недостаточный эффект этих препаратов; ПВН; пирамидные и/или мозжечковые знаки; похолодание, зябкость конечностей; контрактуры; диспропорциональный антеколлис; выраженная дисфония, дисфагия или дизартрия; респираторный стридор; нерегулярный тремор или миоклонии.
Прогрессирующий надъядерный паралич
Болезнь диффузных телец Леви
В последнее время стали выделять новую нозологическую форму, протекающую с синдромом П – болезнь диффузных телец Леви (БДТЛ). Тельца Леви – это внутриклеточные эозинофильные цитоплазматические включения, которые обнаруживаются в клетках черного вещества при БП и считаются маркером этой болезни. При БДТЛ они встречаются не только в черном веществе, но в большом количестве широко диссеминированы по всему головному мозгу. Диагноз БДТЛ – патоморфологический диагноз. Клинически это заболевание характеризуется П, который обычно хорошо лечится препаратами леводопы, наряду с деменцией с выраженными зрительными галлюцинациями. Типичным является также флуктуация выраженности расстройств высших психических функций – в основном за счет изменения способности концентрации внимания.
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) и другие заболевания
Существует несколько более редких причин истинного или псевдопаркинсонизма. Одна из них, о которой следует всегда вспомнить при наличии П у людей моложе 45 лет (в том числе у детей), это гепатолентикулярная дегенерация, или болезнь Вильсона-Коновалова. Это наследственное заболевание, при котором отмечается нарушение метаболизма меди в организме из-за недостаточности фермента церулоплазмина. В результате медь в избыточном количестве откладывается в печени, базальных ганглиях и вокруг радужной оболочки глаза. Болезнь Вильсона-Коновалова следует подозревать не только при наличии П у молодых людей, но и при возникновении у них других признаков поражения экстрапирамидной системы (например, дистонии) или психических расстройств. Диагностика основана на обнаружении с помощью щелевой лампы отложения меди вокруг радужки – кольцо Кайзера–Флейшера. Последнее на стадии неврологических проявлений имеет место у 98% больных. Диагностическое значение имеет также исследование экскреции меди с мочой и концентрации церулоплазмина в крови. Последнее, однако, в отсутствие кольца Кайзера–Флейшера и нормальной экскреции меди не имеет диагностической значимости. Если ситуация продолжает оставаться неясной, проводится биопсия печени или генетическое тестирование. Болезнь Вильсона-Коновалова довольно успешно лечится с помощью D-пеницилламина и препаратов цинка в сочетании с диетой.
Токсический П может иметь место при интоксикации марганцем. Нередко встречается П, вызванный токсином МФТП (1-метил-4-фенил- 1, 2, 3, 6-тетрагидропиридин), так как оно избирательно поражает черную субстанцию, вызывая гибель дофаминовых нейронов. Схожий с этим веществом токсин вырабатывается при кустарном приготовлении некоторых наркотических веществ, поэтому П нередко наблюдается среди молодых людей, страдающих наркоманией. Наряду с П при этом могут отмечаться пирамидные и псевдобульбарные симптомы.
П встречается при энцефалопатии боксеров. Считается, что в таком случае этиологическую роль играют регулярно получаемые множественные черепно-мозговые травмы. Следует помнить, что энцефалопатия боксеров – практически единственная ситуация, когда черепно-мозговые травмы играют роль в развитии П. В целом у людей, не занимающихся боксом, наличию черепно-мозговой трамы в анамнезе (одной или нескольких) не придается этиологическое значение.
П иногда может иметь место при хорее Гентингтона, которое обычно протекает с выраженными гиперкинезами. Это наследственное заболевание обычно дебютирует на четвертом десятилетии жизни, однако встречаются случаи и более раннего начала. Обычно с акинетико-ригидным синдромом протекает так называемая ювенильная форма хореи Гентингтона (форма Вестфаля). Встречаются также семейные случаи П, в том числе ювенильного. Поэтому у всех больных, имеющих синдром П, сбор наследственного анамнеза имеет большое значение.
Акинезия и ригидность могут иногда наблюдаться при болезни Альцгеймера, Пика, при которых ядром клинической картины является прогрессирующая деменция. При болезни Альцгеймера в первую очередь страдает память, а при болезни Пика на первый план выступает распад личности. При этих заболеваниях П чаще проявляется умеренной акинезией, ригидность выражена меньше, а паркинсонический тремор практически никогда не наблюдается.
Постэнцефалитический П в наше время практически не встречается. Много случаев наблюдалось после пандемии летаргического энцефалита в начале XX столетия. С тех пор в литературе описано только несколько случаев.
Особая эндемическая форма П наблюдается на острове Гуам и в некоторых других местах восточной части Тихого океана. В этих местах отмечается много случаев П в сочетании с амиотрофиями и деменцией (гуамский комплекс паркинсонизм – деменция). Патоморфологически мозг этих больных выглядит, как мозг больных ПНП. Одинаковыми являются также гистологические маркеры.
Читайте также: