
Подострая некротизирующая энцефалопатия болезнь лейга

Синдром Лея (подострая некротизирующая энцефалопатия) – это наследственная болезнь, которая проявляется нейрометаболическим синдромом, поражающим центральную нервную систему. Чаще всего этим заболеванием болеют дети до трех лет, гораздо реже оно проявляется у подростков и взрослых.
Болезнь Лея может иметь острое, подострое или хроническое течение. Проявляемые симптомы достаточно выразительные для того, чтобы диагностировать синдром на ранних стадиях. Распространение данной болезни зависит от наследственного фактора, если в семье есть больной человек, то вероятность передачи заболевания детям достаточно высока.
Что провоцирует развитие синдрома
Подострая некротизирующая энцефаломиопатия развивается вследствие нехватки ферментов, отвечающих за формирование энергетического обмена в организме.
Недостача ферментов возникает на фоне неправильного метаболизма пировиноградной кислоты и дефектов миграции электронов в респираторные цепи. Также синдром Ли возникает по следующим причинам:
- генетическая детерминанта;
- наличие новообразований в мозжечке, глиоз и пролификация сосудов;
- компликация дегенеративных заболеваний ЦНС;
- мутация генов на фоне неправильной работы митохондрий;
- воспалительные процессы в сосудах;
- отмирание нейронов и структурных частиц мозга;
- ранее перенесенные инфекции и вакцинации.
Формы протекания болезни
Для синдрома Лея свойственно чаще всего подострое или хроническое протекание, которое влечет за собой смертельный исход. Гораздо реже 
болезнь протекает по форме острой энцефалопатии.
При стремительной форме развития заболевания настает апоплексия респираторного центра, что приводит к смерти.
Существуют мягкие формы течения болезни, сопровождающиеся замедлением психомоторного онтогенеза.
При любой форме болезни, протекание ее вариабельно. После стремительного клинического ухудшения наступают кратковременные улучшения.
Симптоматика синдрома
Для каждой формы болезни Лея характерна своя клиническая картина. Подострая форма энцефаломиопатии проявляется следующими признаками:
- мышечная слабость, переходящая в гипертонус;
- общая слабость организма;
- признаки пирамидной и экстрапирамидной дефицитности;
- молочно-кислый ацидоз;
- периодические вспышки миоклонии;
- кардиомиопатия;
- атаксия и тремор конечностей;
- гиповентиляция;
- понижение сухожильных рефлексов.
При быстрой и прогрессирующей форме протекания синдрома возникают такие симптомы:
- клонические и тонические судороги;
- атаксия;
- омертвение зрительного нерва;
- спастические парезы;
- редкие эпилептические приступы;
- центральная скотома;
- стремительное слабоумие.
Общая клиническая характеристика для всех форм синдрома Лея:
- недомогание, тошнота и рвота;
- сонливость и пониженная внимательность;
- падение аппетита или полное его отсутствие;
- быстрая утомляемость и нарушение сознания;
- прогрессирующее убавление массы тела;
- инволюция зрительного нерва;
- расстройство координации;
- дистрофическая миотония;
- дрожание конечностей;
- неправильный акт глотания.

Комплекс диагностических процедур
Для установления диагноза Лея применяются следующие методы исследований и берутся такие анализы:
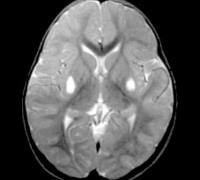
- МРТ головного мозга. Данный вид исследования позволяет выявить характерные для болезни нарушения: поражение ствола мозга с обеих сторон, пертурбация ткани головного мозга, появление липидных включений.
![]()
- ЭКГ. Определяется деривация (смещение) электрической оси вправо. Также выявляется синусовая тахикария и понижение вольтажа.
- Биохимическое исследование крови. Определяется наличие лактата-ацидоза (из-за накопления в крови обильного количества молочной и пировиноградной кислоты). С помощью данного исследования выявляется повышенный уровень кетоновых тел.
- Проведение электроэнцефалографии. По ее результатам определяются фокальные проявления эпилептической активности.
- Морфологические исследования. Обнаруживаются изменения вещества мозга: страдают таламус, подкорковые узлы, зрительный нерв, выявляются симметричные очаги некроза.

Помимо вышеперечисленных методов диагностики, проводятся и вспомогательные исследования:
- Осмотр окулиста. Выявляется отмирание зрительного нерва и конгенитальное недоразвитие.
- Осмотр кардиолога и кардиохирурга. При проведении этого осмотра можно определить увеличение ствола легочной артерии и дефицитность двухстворчатого клапана.
- Электромиография. Устанавливается миопатичекоое поражение двигательной единицы.
Лечение и прогноз

Лечение некротизирующей энцефалопатии не всегда возможно. Если заболевание переходит в хроническую форму, то летальный исход неизбежен.
Целью лечения является улучшение состояния центральной нервной системы. Для этого назначаются медикаменты, в состав которых входит витамин В1. Также назначаются антибиотики, лучшими из которых являются Ампициллин и Биотин. При медикаментозной терапии необходимо соблюдать диету, при которой можно употреблять не более 1 грамма белка в сутки.
В группе риска находятся лица, имеющие наследственную предрасположенность к данной болезни и дети, возрастом до трех лет. При не предоставлении медицинской помощи, болезнь заканчивается смертельным исходом. Средняя продолжительность жизни с момента заболевания составляет пять лет.
Профилактические меры
Профилактика заболевания заключается в своевременной терапии болезней ЦНС и заболеваний, которые оказывают влияние на центральную нервную систему. Также необходимо рациональное питание и вести здоровый образ жизни.
Действенность данного метода профилактики доказана в конце 2016 года и пока что на практике почти не используется.

Общие сведения
Синдром Лея относится к редким нейрометаболическим синдромам, поражающим ЦНС, и вызывающим нарушение координации движений и мышления и летальный исход. Заболевание наследственное по аутосомно-рецессивному типу или Х-сцепленному. Патологии чаще всего подвержены маленькие детки до 2-х лет – 1-2 ребенка на десятки тысяч, в более редких случаях – подростки и взрослые особы. Код по МКБ-10: G 31.8
Миру стало известно об этом заболевании благодаря Денису Лею, который описал его еще в 1951 году. В научных кругах обособить синдром от подобных энцефалопатий удалось в 1954 г. Связь с митохондриальной активностью смогли выявить 1968 г, но главным продвижением стало обнаружение мутации в цитохромоксидазах в 1977 г.
Проблема состоит в том, что эффективных препаратов или методов лечения болезни Лея до сих пор не разработано, ведь причин её множество, начиная с влияния мутаций в генах, отвечающих за работу митохондрий, и заканчивая – осложнениями дегенеративных заболеваний нервной системы, аномальными образованиями некротизированных очагов или прорастания сосудов, глиоза в структурах головного мозга.
Патогенез
Для таких митохондриальных заболеваний как синдром Лея характерно полиорганное поражение с вовлечением нервных и мышечных тканей. В основе патологии обычно лежит сбой регулирования обмена пировиноградной кислоты, при этом наблюдается нарушение обменных реакций, снижение выработки энергии и замедление процессов перемещения электронов в клетках дыхательной системы. Процессы могут спорадически усилиться, чему способствует наследственные факторы. Для запуска развития синдрома необходимо присутствие в организме более 90% мутантной мтДНК от всей мтДНК. В случае меньшего содержания мутантной ДНК симптоматика напоминает нейропатию и сводится к атаксии и пигментному ретиниту.
Для болезни характерно раннее развитие и стремительное злокачественное течение с присоединением большого количества различных неврологических нарушений, затем начинаются проблемы с дыханием и метаболизмом.
Классификация
В зависимости от патогенеза и возрастных особенностей выделяют различные варианты нарушений, выражающиеся в виде:
- недостаточности цитохром С-кислород-оксидоредуктазы;
- некротизирующей подострой инфантильной энцефалопатии Лея;
- синдрома Лея у взрослых особ;
- синдрома NARP или дефекта митохондриальной ДНК;
- Х-сцепленного синдрома Лея;
- недостаточности фермента – пируваткарбоксилазы.
В зависимости от течения синдром Лея бывает:
- острой формы – в очень редких случаях и напоминает острую энцефалопатию;
- хроническая и подострая болезнь Лея.
Причины
Главным провоцирующим фактором синдрома Лея принято считать недостаточность цитохромоксидазы и мутации генов, отвечающих за работу митохондрий:
- замену Т (тимина) на Г (гуанин) или Ц (цитозин) в 8933-м положении 6-й субъединицы АТФ-синтазы;
- мутации в флавопротеиновой единице сукцинатдегидрогеназного комплекса, в SURF-генах либо в двух ядерных генах (приводит к развитию митохондриальных болезней второго класса), в целом – их описано около 40.
Однако, способствовать нейрометаболической энцефалопатии может:
- образование некротизированных очагов или онкоструктур (злокачественных либо доброкачественных) в структурах мозжечка или мозгового ствола;
- прорастания сосудов или развитие глиоза;
- наследственная предрасположенность и мутагенные факторы любой природы – биологические, экологические, химические и пр.;
- осложнение дегенеративных заболеваний, поражающих нервную систему;
- а также результате вакцинации.
Симптомы
Синдром Лея и его прогрессирование приводит:
- к возникновению тошноты и рвоты;
- к быстрому снижению массы тела и слабости;
- к развитию задержки психомоторных функций (например, снижению внимательности) и нарушениям сознания;
- к приступам тонико-клонических судорог либо мышечной дистонии/гипотонии;
- к возникновению респираторных аномалий;
- к атрофии зрительных нервов, возможно даже слепоте;
- к тугоухости;
- к повышению утомляемости и сонливости;
- а также к нарушениям координации, сухожильных рефлексов, актов глотания, тремору конечностей.
Клиническая характеристика и патогенез схожи, но симптоматика может дополняться:
- метаболическим компенсаторным ацидозом или приступами тяжелого ацидоза;
- снижением уровня бикарбонатов в крови;
- увеличением объемов молочной, пировиноградной кислоты в крови и ликворной жидкости;
- приступами миоклонии;
- гиповинтиляциейи развитием тахиапноэ;
- атаксиейи клоническими подергиваниями;
- спастическими парезами;
- эпилептическими припадками;
- прогрессирующей деменцией;
- центральной скотомой;
- кардиомиопатией;
- симптомами экстрапирамидной и пирамидной недостаточности.
Анализы и диагностика
Болезнь Лея относится к подострым либо хроническим некротизирующим энцефаломиопатиям и является важным направлением изучения в детской невралгии. Для её диагностики необходима консультация у врача-невролога, а также:
- проведение общего анализа крови и её биохимических исследований, выявляющих лактатацидоз(повышенную концентрацию лактата, пирувата в кровяном русле и ликворе), снижение количества карнитина, активности цитохромоксидазы в культуральных клетках — фибробластах;
- изучение структур головного мозга посредством магнитно-резонансной томографии;
- изучение электрической активности (ЭЭГ) и морфологических структур головного мозга.
Лечение
Медикаментозное консервативное лечение болезни Лея обычно симптоматическое и включает использование:
- антибиотиков, например, Ампициллина;
- различных методов диализа;
- ноотропов;
- противосудорожных средств;
- препаратов, содержащих В1 и биотин.
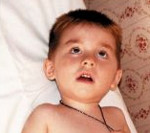
Синдром Ли – генетически гетерогенное наследственное заболевание, характеризующееся разнообразными нарушениями метаболизма и формирования компонентов центральной нервной системы. Симптомы этой патологии, как правило, проявляются еще в раннем детстве, к ним относят мышечную гипотонию, проблемы со вскармливанием и задержку психомоторного развития. При дальнейшем прогрессировании заболевания возникают эпилептические припадки, гиперкинезы, дыхательные расстройства. Диагностика синдрома Ли осуществляется на основании данных настоящего статуса больного, магнитно-резонансной томографии, молекулярно-генетических анализов. Специфического лечения данной патологии не существует, симптоматическая терапия лишь незначительно замедляет прогрессирование заболевания.
- Причины и классификация синдрома Ли
- Симптомы
- Диагностика и лечение синдрома Ли
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Синдром Ли (подострая некротизирующая энцефаломиелопатия) – наследственное нейродегенеративное заболевание центральной нервной системы, которое характеризуется ранним началом и неуклонным прогрессированием неврологических нарушений. Впервые данное состояние было описано в 1951 году английским психиатром Денисом Ли, который определил его как наследственный вариант энцефаломиелопатии. Дальнейшие исследования показали, что синдром Ли является крайне гетерогенным состоянием с точки зрения этиологии – его причиной становятся дефекты множества генов, расположенных на аутосомах, Х-хромосоме и митохондриальной ДНК. По этой причине механизм наследования заболевания может быть (в зависимости от характера мутации) аутосомно-рецессивным, сцепленным с полом или митохондриальным. Из-за разнообразия генетических дефектов, являющихся причиной синдрома Ли, различается и половое распределение этого состояния, однако, по мнению многих врачей-генетиков, в целом можно считать, что оно в равной степени поражает как мальчиков, так и девочек. Встречаемость составляет ориентировочно 1 случай на 34-36 тысяч новорожденных.
Причины и классификация синдрома Ли
Причинами развития синдрома Ли могут выступать мутации широкого спектра генов, расположенных на разных хромосомах. Однако патогенез этого состояния примерно сходен у различных форм патологии и чаще всего связан с нарушением процессов клеточного дыхания и функционирования дыхательной цепи митохондрий. В отношении некоторых форм синдрома Ли также замечено нарушение функционирования пируватдегидрогеназного комплекса. Нарушение структуры белков дыхательной цепи митохондрий приводит к недостаточному синтезу АТФ, являющемуся основным источником энергии во всех клетках организма. Нейроны и клетки нейроглии особенно чувствительны к недостатку энергии, что становится причиной развития разнообразных нарушений еще с детского возраста. Классификация всех генетических дефектов при синдроме Ли основана на том, какой компонент дыхательной цепи (представляющей собой белковый комплекс) митохондрий нарушен в результате мутации.
- Синдром Ли, обусловленный поражением комплекса 1 (НАДН-KoQ-редуктаза). Этот вариант может наследоваться как аутосомно-рецессивно, так и митохондриально. Наиболее распространенные варианты заболевания этого типа обусловлены мутациями ядерных генов NDUFA10 (расположен на 19-й хромосоме), NDUFS4 и DUFAF2 (5-я хромосома), NDUFS3 (11-я хромосома). Кроме того, к развитию синдрома Ли в результате поражения митохондриального комплекса 1 способны приводить дефекты митохондриальной ДНК – генов MTND1, MTND2 и MTND3. Результатом этого является нарушение начального этапа переноса электронов и водорода в дыхательной цепи, что снижает синтез АТФ.
- Синдром Ли, вызванный дефектами белков, входящих в митохондриальный комплекс 2 (сукцинат-KoQ-редуктаза). Заболевание этого типа наследуется только аутосомно-рецессивно, достоверно удалось установить взаимосвязь между синдромом Ли и мутациями гена SDHA, локализованного на 5-й хромосоме. Данный ген кодирует субъединицу А сукцинатдегидрогеназного комплекса, при генетических нарушениях такого типа активность фермента снижается, что и ведет к развитию заболевания.
- Синдром Ли, возникающий в результате нарушения структуры белков митохондриального комплекса 3 (KoQН2-цитохром с-редуктаза). К этой разновидности относят наиболее распространенный вариант заболевания, вызванный мутацией гена BCS1L, расположенного на 2-й хромосоме. Развитие синдрома Ли обусловлено пониженной экспрессией фермента убихинон-с-редуктазы, входящего в состав митохондриального комплекса 3. Его выделение регулируется специфическим белком, который кодируется геном BCS1L – в результате мутации полученный дефектный протеин не способен полноценно выполнять свои функции. Для этого варианта синдрома Ли характерно аутосомно-рецессивное наследование.
- Синдром Ли, обусловленный повреждением митохондриального комплекса 4 (цитохром с-оксидаза). Может быть вызван как мутациями ядерных генов (COX10, SCO1), в основном расположенных на 17-й хромосоме, так и повреждением митохондриальной ДНК – это удалось выяснить по характеру наследования некоторых форм, однако ключевые гены пока не определены.
- Синдром Ли, вызванный нарушением структуры митохондриального комплекса 5 (АТФ-синтаза). К этому варианту относят сравнительно редкие мутации гена ATPAF2, локализованного на 17-й хромосоме. В результате мутации нарушается работа АТФ-синтазы, образование АТФ окислительным путем резко снижается.
Симптомы
Проявления синдрома Ли обычно возникают на протяжении первого года жизни ребенка, иногда они могут регистрироваться в возрасте 2-5 лет, в редких случаях развитие заболевания начинается в подростковый период. Обычно первым проявлением патологии становится сонливость или, наоборот, повышенная возбудимость ребенка, у грудных детей наблюдается нарушение питания, недобор массы тела. В дальнейшем синдром Ли приводит к задержке психофизического развития, а у детей старшего возраста – к постепенной утрате уже обретенных навыков. Среди других неврологических симптомов заболевания наиболее часто отмечаются парезы, тремор конечностей, нарушение координации движения, поражение периферических нервов, снижение сухожильных рефлексов. В дальнейшем могут регистрироваться клонические судороги и эпилептические припадки.
Из-за недостатка энергии, обусловленного синдромом Ли, страдает не только нервная система, но и другие органы с высоким потреблением АТФ. В большинстве случаев у больных детей отмечается мышечная гипотония и слабость. Затрагивает заболевание и печень – орган с очень высоким потреблением энергии. У пациентов с синдромом Ли нередко выявляется увеличение печени, желтуха, иногда гепатолиенальный синдром. По мере прогрессирования патологии возникают нарушения дыхания – оно становится затрудненным, иногда приобретает характер дыхания Чейна-Стокса. У ряда больных со временем развивается миокардиодистрофия.
Синдром Ли имеет прогрессирующие течение. На терминальных этапах наблюдается поражение органов зрения, которое проявляется нистагмом, нарушением цветовосприятия, косоглазием. В конечном итоге может возникнуть атрофия зрительного нерва и полная слепота. Мышечная гипотония и гипорефлексия сменяются спастическим напряжением мышц и повышением сухожильных рефлексов. Через 2-7 лет после появления первых симптомов синдрома Ли происходит резкое падение массы тела, вышеперечисленные проявления резко усиливаются, наступает летальный исход по причине дыхательной или сердечно-сосудистой недостаточности.
Диагностика и лечение синдрома Ли
Для определения наличия синдрома Ли применяют магнитно-резонансную томографию головного мозга, электронейромиографию, изучение наследственного анамнеза, молекулярно-генетические анализы. При осмотре выявляют характерные неврологические симптомы, тремор конечностей, отставание в психофизическом развитии, у младенцев – недобор массы тела. На магнитно-резонансной томографии мозга обнаруживают симметричные изменения в области продолговатого мозга, таламуса и моста, иногда аналогичные изменения могут регистрироваться и в спинном мозге. Наилучшие результаты диагностики синдрома Ли при помощи МРТ получаются при использовании T2W и FLAIR режимов.
В тех случаях, когда имеются признаки поражения периферических нервов и мышц, для диагностики синдрома Ли выполняют электронейромиографию. При этом заболевании главным и наиболее частым результатом ЭНМР становится замедление скорости прохождения нервного импульса, которое свидетельствует о демиелинизации нервов. Изучение наследственного анамнеза информативно в случае аутосомно-рецессивных форм заболевания, при мутации генов митохондриальной ДНК четко определить семейный характер патологии затруднительно. Молекулярно-генетическая диагностика массово используется только в отношении некоторых форм синдрома Ли (обусловленных мутациями генов BCS1L, SURF1 и некоторых других).
Специфического лечения данной патологии не существует, применяется симптоматическая терапия: противосудорожные и ноотропные средства, препараты для улучшения мозгового кровообращения. Важную роль в лечении синдрома Ли играет назначение витаминов, служащих кофакторами ферментов дыхательной цепи митохондрий – В1, В6, Q10. Их регулярный прием позволяет несколько замедлить прогрессирование заболевания и уменьшить выраженность симптомов. Однако, несмотря на все предпринятые терапевтические меры, 80% больных умирает через 2-7 лет после регистрации первых проявлений патологии.
Прогноз и профилактика
Прогноз синдрома Ли крайне неблагоприятный, так как большинство больных умирает через несколько лет после возникновения заболевания. Симптоматическое лечение может несколько замедлить прогрессирование патологии и ослабить выраженность проявлений, однако полноценного улучшения оно не обеспечивает. В большинстве случаев еще за год-два до летального исхода наступает полная инвалидизация больного, обусловленная неврологическими, дыхательными и метаболическими нарушениями. Причиной смерти при синдроме Ли чаще всего становится сердечно-сосудистая или дыхательная недостаточность. Профилактика этого заболевания осуществляется в рамках медико-генетического консультирования родителей перед зачатием ребенка.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Причины
- Симптомы
- Диагностика
- Как обследовать?
- Какие анализы необходимы?

О заболевании впервые было упомянуто в 1951 г. К настоящему времени описано более 120 случаев. Болезнь Лея (OMIM 256000) - генетически гетерогенное заболевание, которое может наследоваться как по ядерному типу (аутосомно-рецессивно или сцепленно с Х-хромосомой), так и митохондриально (реже).

[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Причины синдрома Лея
В основе заболевания лежит дефицит ферментов, обеспечивающих образование энергии главным образом за счёт нарушения обмена пировиноградной кислоты и дефекта транспорта электронов в дыхательной цепи. Развивается дефицит пируватдегидрогеназного комплекса (а-Е1-субъединицы), пируваткарбоксилазы, комплекса 1 (НАД-коэнзим Q-редуктаза) и комплекса 4 (цитохромоксидаза) дыхательной цепи.
При этом установлено, что дефекты пируваткарбоксилазы, комплекса 1 (НАД-коэнзим Q-редуктаза) и комплекса 4 (цитохромоксидаза) дыхательной цепи наследуются по аутосомно-рецессивному типу, дефекты пируватдегидрогеназного комплекса (а-Е1-субъединицы) - Х-сцепленно рецессивно. При точковых мутациях мтДНК, которые затрагивают 6-ю субъединицу АТФазы, характерно митохондриальное наследование. Чаще всего происходит мисценс-мутация, связанная с заменой тимина на гуанин или цитозин в положении 8993 мтДНК. Реже встречается мутация в позиции 9176 мтДНК. В связи с тем что мутация T8993G - основной дефект при синдроме NARP, описаны семьи с наличием этих двух заболеваний. У детей описана также мутация мтДНК в позиции 8344, которая встречается при синдроме MERRF.
Предполагают, что в случае накопления мутантной мтДНК в большинстве митохондрий развивается тяжёлое течение синдрома Лея. При митохондриальном генезе этого состояния мутантную мтДНК обнаруживают в 90% всех митохондрий. Патогенез связан с нарушением образования энергии в клетках и развитием лактат-ацидоза.
[7], [8], [9], [10], [11], [12], [13], [14]
Симптомы синдрома Лея
Первые признаки заболевания дебютируют в раннем возрасте (1-3 года). Однако известны случаи манифестации болезни в 2-недельном и в 6-7-летнем возрасте. Вначале развиваются неспецифические нарушения: задержка психомоторного развития, снижение аппетита, эпизоды рвоты, дефицит массы тела. В последующем нарастают неврологические симптомы: мышечная гипотония или дистония с переходом в гипертонус, приступы миоклонии или тонико-клонические судороги, тремор конечностей, хореоатетоз, расстройство координации, снижение сухожильных рефлексов, вялость, сонливость. Церебральная нейродегенерация носит прогрессирующий характер. Нарастают симптомы пирамидной и экстрапирамидной недостаточности, нарушается акт глотания. Нередко наблюдают такие изменения органа зрения, как птоз, офтальмоплегия, атрофия зрительных нервов, реже пигментная дегенерация сетчатки. Иногда развиваются гипертрофическая кардиомиопатия, появляются эпизоды тахипноэ.
Редко заболевание протекает по типу острой энцефалопатии. Более характерно хроническое или подострое течение, которое приводит к летальному исходу через несколько лет после начала заболевания. При быстром течении (несколько недель) смерть наступает в результате паралича дыхательного центра.
[15], [16], [17], [18], [19], [20], [21]
Диагностика синдрома Лея
При биохимическом исследовании крови выявляют лактат-ацидоз вследствие накопления молочной и пировиноградной кислот в крови и ликворе, а также увеличение содержания аланина в крови. Также может быть повышен уровень кетоновых тел. В моче выявляют повышенную экскрецию органических кислот: молочной, фумаровой и др. Часто снижается уровень карнитина в крови и тканях.
По результатам ЭЭГ выявляют фокальные признаки эпилептической активности. По данным МРТ обнаруживают расширение желудочков мозга, двустороннее поражение мозга, кальцификацию базальных ганглиев (хвостатого ядра, скорлупы, чёрной субстанции, бледного шара). Можно также выявить атрофию больших полушарий и вещества мозга.
При морфологическом исследовании обнаруживают грубые изменения вещества мозга: симметричные очаги некроза, демиелинизации и губчатой дегенерации мозга, преимущественно средних отделов, моста, подкорковых узлов, таламуса, зрительного нерва. Гистологическая картина включает кистозное перерождение мозговой ткани, астроцитарный глиоз, гибель нейронов, увеличение количества митохондрий в клетках. В скелетных мышцах - накопление липидных включений, снижение гистохимической реакции на комплексы 1, 4 дыхательной цепи, субсарколеммальное скопление митохондрий, аномальные митохондрии с дезорганизацией крист. Феномен RRF часто не обнаруживают.
Читайте также: