
С чем дифференцировать болезнь паркинсона

Синдром паркинсонизма складывается из следующих клинических симптомов: различного сочетания гипокинезии, ригидности, тремора покоя и постуральных расстройств.
Под гипокинезией в настоящее время подразумевают снижение инициации движения и способности продолжать движение с нормальной скоростью. В рамках гипокинезии различают два основных феномена:
- брадикинезию, т. е. собственно снижение скорости движения;
- олигокинезию, проявляющуюся затруднением начала движения и значительным упрощением двигательного рисунка в виде отсутствия содружественных синкинезий (как частный вариант – ахейрокинез, проявляющийся отсутствием содружественных движений рук при ходьбе), расстройством выполнения последовательных движений, фрагментацией движений.
Наиболее простой пробой для выявления гипокинезии в верхних конечностях является оценка движений пальцев рук: пациента просят сводить и разводить большой и указательный пальцы кисти с максимальной скоростью, при этом оценивается характер начала движения, скорость, симметричность, амплитуда.
К наиболее классическим симптомам гипокинезии относят гипомимию – маскообразное лицо, гипофонию, микрографию, слюнотечение (нарушение сглатывания слюны). Следует подчеркнуть, что для выявления синдрома паркинсонизма решающее значение имеет обнаружение именно гипокинезии.
Тремор часто наблюдается при паркинсонизме и также часто является причиной ошибочной диагностики этого синдрома, так как принимается основным клиническим проявлением заболевания. Это наиболее заметный симптом, именно на него в первую очередь обращают внимание пациент и окружающие.
В классическом варианте при болезни Паркинсона дрожание носит характер так называемого тремора покоя, с частотой 5 ± 2 Гц, который исчезает при целенаправленном действии, преимущественно вовлекаются дистальные отделы конечностей и нижняя челюсть. В связи с этим важно оценивать тремор в состоянии покоя (например, когда руки пациента лежат на коленях), при удержании положения (руки вытянуты вперед) и при целенаправленных действиях (пальценосовая проба).
Для оценки простуральной устойчивости врач становится позади пациента и подталкивает его за плечи на себя, в норме для удержания равновесия человек иногда делает до двух шагов.
Классификация паркинсонизма
Таким образом, на первом этапе диагностики основным является выявление синдрома паркинсонизма, для которого облигатным симптомом является наличие гипокинезии с различным сочетанием ригидности, тремора и постуральных нарушений.
На втором этапе уточняют нозологическую форму. В настоящее время паркинсонизм подразделяют на следующие группы заболеваний:
- Первичный (идиопатический) паркинсонизм:
- болезнь Паркинсона,
- юношеский (ювенильный) паркинсонизм.
- Вторичный (симптоматический) паркинсонизм (вследствие поражения головного мозга определённой этиологии):
- сосудистый паркинсонизм,
- токсические энцефалопатии (интоксикации марганцем, ртутью),
- гидроцефалия,
- посттравматический паркинсонизм,
- опухоли,
- постэнцефалитический паркинсонизм,
- метаболические энцефалопатии.
- Паркинсонизм при других дегенеративных и наследственных заболеваниях (паркинсонизм-плюс):
- прогрессирующий надъядерный паралич (болезнь Стила-Ричардсона-Ольшевского),
- мультисистемная атрофия,
- болезнь диффузных телец Леви,
- кортико-базальная дегенерация,
- паркинсонизм-деменция-БАС,
- гемипаркинсонизм-гемиатрофия,
- болезнь Генгингтона,
- идиопатическая (семейная) кальцификация базальных ганглиев (болезнь Фара),
- гепатолентикулярнал дегенерация (болезнь Вильсона-Коновалова) и т. д.
При этом необходимо учитывать, что по частоте встречаемости среди всей популяции пациентов, страдающих паркинсонизмом, болезнь Паркинсона составляет 70-80 %, вторичный паркинсонизм – 10-15 %, паркинсонизм-плюс – 10-15 %. Рассмотрим особенности отдельных, наиболее часто встречающихся нозологических форм.
Первичный (идиопатический) паркинсонизм
По данным ВОЗ, в мире насчитывается около 3,7 млн (0,06 % населения) людей с БП. Средний возраст начала заболевания составляет 55 ± 10 лет. В большинстве исследований соотношение числа мужчин и женщин с БП колеблется от 1,1 до 1,6. Распространённость БП в структуре общей популяции колеблется, по данным разных авторов, от 60 до 187 человек на 100 тыс. населения.
В последнее время в развитых странах отмечается некоторый рост заболеваемости БП, что связывают с увеличением средней продолжительности жизни населения, а также с улучшением диагностических возможностей современной медицины.
Несмотря на большое число исследований, направленных на поиск основного фактора развития БП, этиология данного заболевания до настоящего времени не известна.
Диагностика болезни Паркинсона
В настоящее время диагноз болезни Паркинсона ставится на основе характерной клинической симптоматики заболевания, т. е. является клиническим. В частности, достаточно широко используют клинико-диагностические критерии Банка головного мозга общества БП Великобритании:
- Синдром паркинсонизма:
- наличие гипокинезии (замедленность инициации произвольных движений с прогрессирующим снижением скорости и амплитуды повторных движений);
- наличие по меньшей мере одного из следующих симптомов: мышечной ригидности, тремора покоя 4-6 Гц, постуральной неустойчивости, не связанной со зрительной, вестибулярной, мозжечковой пли проприоцептивной дисфункцией.
- Критерии исключения болезни Паркинсона:
- наличие в анамнезе повторных инсультов со ступенеобразным прогрессированием симптомов паркинсонизма, повторные черепно-мозговые травмы или достоверный энцефалит;
- кулогирные кризы;
- лечение нейролептиками перед дебютом болезни;
- длительная ремиссия;
- строго односторонние проявления в течение более трёх лет;
- супрануклеарный паралич взора;
- мозжечковые знаки;
- раннее появление симптомов выраженной вегетативной недостаточности;
- раннее появление выраженной деменции;
- симптом Бабинского;
- наличие церебральной опухоли или открытой (сообщающейся) гидроцефалии;
- негативная реакция на большие дозы Л-ДОФА (если исключена мальабсорбция);
- интоксикация МФТП.
- Критерии, подтверждающие БП (для достоверного диагноза необходимо наличие трёх и более симптомов):
- одностороннее начало проявлений болезни;
- наличие тремора покоя;
- постоянная асимметрия с более выраженными симптомами на стороне тела, с которой началась болезнь;
- хорошая реакция (70-100 %) на Л-ДОФА;
- прогрессирующее течение заболевания;
- наличие выраженной дискинезии, индуцированной Л-ДОФА;
- откликаемость на Л-ДОФА в течение пяти лет и более;
- длительное течение заболевания (10 лет и более).
Таким образом, клиническая диагностика болезни Паркинсона носит трёхступенчатый характер. На первом этапе выявляют синдром паркинсонизма по наличию гипокинезии в различном сочетании с тремя другими кардинальными симптомами заболевания. На втором этапе проводят дифференциальную диагностику с вторичным паркинсонизмом и паркинсонизмом-плюс, для чего выявляют уже перечисленные характерные особенности, которые мы рассмотрим ниже. На третьем этапе выявляют признаки, подтверждающие диагноз БП.
В клинически развёрнутой стадии постановка диагноза БП не вызывает особых сложностей у грамотного специалиста. Однако клинические симптомы БП появляются при гибели 50-80 % дофаминергических нейронов чёрной субстанции.
В рамках появляющихся новых данных о наличии у ряда современных лекарственных средств нейропротективного действия в отношении дофаминергических нейронов, а также поиска новых методов патогенетического лечения данного заболевания представляется перспективным разработка ранних методов диагностики БП, что позволит осуществлять более эффективное терапевтическое воздействие на фоне сохранности большего числа дофаминергических нейронов. В связи с этим рассмотрим принципиальные возможности ранней диагностики данного заболевания исходя из имеющихся в настоящее время данных о патогенезе БП.
Обнаружение генетических мутаций при БП и как следствие нарушение обмена ряда белков, например, альфа-синуклеина, стимулировало поиск генетических маркёров при БП. Однако обнаруженные в настоящее время маркёры имеются лишь у части пациентов с клиническим диагнозом БП.
На биохимическом уровне имеется дефект активности митохондриального комплекса I, оценка активности которого в тромбоцитах может применяться в качестве биохимического маркёра БП. Также предложено определять уровень тирозин-гидроксилазы, дофамина и рецепторов дофамина в лимфоцитах периферической крови, уровень которых в данных клетках крови, по данным ряда авторов, снижается уже при начальных проявлениях БП.
Рядом авторов предлагается оценивать степень поражения чёрной субстанции по уровню железа с помощью транскраниальной ультрасонографии. При этом следует отметить, что обнаруженные нейробиохимические изменения непосредственно в дофаминергических нейронах чёрной субстанции, такие как дефицит глутатиона, накопление железа, дефицит цинка, не являются специфичными только для БП, а также распространены не у всех пациентов с БП.
Патоморфологические изменения, например, тельца Леви, возможно регистрировать лишь гистохимически, что не позволяет в современных условиях проводить прижизненную идентификацию данных морфологических изменений. Диагностическую ценность также снижает то, что данные тельца обнаруживаются не у всех пациентов с БП.
Наиболее перспективными в прижизненной оценке структурно-функционального состояния церебральных нейротрансмиттерных систем являются методы функциональной нейровизуализации, такие как позитронно-эмиссионная томография (ПЭТ) и однофотонная эмиссионная компьютерная томография (SPECT). Эти методики позволяют прижизненно изучать функциональное состояние, например, обмена дофамина в структурах головного мозга.
С помощью ПЭТ проводился анализ скорости прогрессирования дегенеративного процесса, и, в частности, было показано, что при БП в среднем накопление (1SF) снижается в хвостатом ядре на 3 %, а в скорлупе на 9 %, что позволило рассчитать продолжительность доклинической стадии при БП – 6 ± 3 года. Основным недостатком методов функциональной нейровизуализации является их высокая себестоимость, что ограничивает их широкое применение в клинической практике.
Определённую информацию можно получить, оценивая плотность дофаминовых рецепторов. В частности, показано, что на начальных стадиях плотность постсинаптических D1-рецепторов не меняется, но отмечается увеличение плотности D2-peцепторов, что отражает механизмы компенсации в условиях дефицита дофамина. На поздних стадиях в большей степени уменьшается плотность D1-peцепторов при относительной сохранности D2-peцепторов, плотность которых остаётся дольше неизменной.
Рядом авторов показана информативность оценки обоняния на ранней стадии БП, в частности они обнаружили снижение способности к различению запахов уже при дебюте данного заболевания.
Показана информативность исследования биоэлектрической активности мышц и относительный анализ параметров поверхностной (накожной) электромиограммы (ЭМГ) при БП. На основании этих данных разработаны количественные характеристики ЭМГ, позволяющие оценивать в первую очередь тремор и ригидность, в меньшей степени – гипокинезию, что можно использовать для оценки эффективности фармакотерапии.
Для объективизации постуральных расстройств применяют методы статической стабилометрии, что позволяет оценить различные аспекты механизма сохранения центра тяжести в площади опоры.
Изменения саккадических движений глаз (СДГ) при БП показаны в ряде исследований, причём большинство из них – зарубежные. В частности, в нашей работе было показано, что при БП имеются достоверные изменения параметров СДГ: латентных периодов, времени перемещения взора и мультисаккадности (р
Дифференциальную диагностику болезни Паркинсона проводят чаще всего с сосудистым, лекарственным, токсическим и постэнцефалитичесим паркинсонизмом; эссенциальным тремором; деменцией с тельцами Леви; прогрессирующим надъядерным параличом; мультисистемной атрофией; гепатолентикулярной дегенерацией.
Сосудистый паркинсонизм. Частота встречаемости сосудистого генеза паркинсонизма, по данным литературы составляет 6-8%. Для диагностики сосудистого паркинсонизма недостаточно иметь в анамнезе сосудистую патологию, даже при наличии характерной клинической картины и данных параклинических исследований. Наличие цереброваскулярной патологии еще в конце прошлого века определяло постановку диагноза сосудистого паркинсонизма, но по данным исследований последних лет необходимо учитывать ее сочетание со следующими критериями:
1) выявление по данным нейровизуализации изменений в стратегически значимых для паркинсонизма зонах (диффузный лейкоареоз; множественные очаги в базальных ганглиях; очаги в таламусе, среднем мозге или лобных долях);
2) малую эффективность препаратов леводопы;
3) наличие в клинической картине постуральной неустойчивости, преимущественного поражения нижних конечностей (апраксия ходьбы), псевдобульбарного синдрома, наличие мозжечковой или пирамидной симптоматики;
4) острое или подострое развитие симптоматики вскоре после перенесенного инсульта с последующей стабилизацией состояния;
5) более выраженные и быстро развивающиеся когнитивные расстройства, чем при болезни Паркинсона.
Лекарственный паркинсонизм. Для лекарственного паркинсонизма характерно:
1) связь с приемом лекарственных препаратов (чаще ими являются нейролептики);
2) подострое развитие заболевания;
3) в дебюте двухстороннее поражение конечностей;
4) наличие смешанного тремора (постурального и покоя);
5) в большинстве случаев регресс симптоматики после отмены соответствующего препарата.
Постэнцефалитический паркинсонизм. Для постэнцефалитического паркинсонизма характерно:
1) наличие в анамнезе энцефалита или инфекции с острым началом и неврологической симптоматикой;
2) более молодой возраст (до 40 лет), чем другие формы паркинсонизма;
3) окулогирные кризы (тоническая судорога взора);
4) глазодвигательные расстройства (обратный синдром Аргайла-Робертсона);
5) вегетативные нарушения (гиперсаливация, гипергидроз и др.);
6) возможны гиперкинезы (спастическая кривошея, мышечная дистония, блефароспазм, тики, оральные гиперкинезы);
7) непрогрессирующее или медленно прогрессирующее течение;
8) наличие в анамнезе гиперсомнического синдрома.
Токсический паркинсонизм. Для токсического паркинсонизма характерно:
1) грубое крупноамплитудное постуральное дрожание конечностей, головы, реже туловища.
2) быстрое нарастание постуральной неустойчивости и нарушений функций ходьбы с частыми падениями, застываниями, тенденцией к ретропульсиям.
3) быстро нарастающий псевдобульбарный синдром с развитием грубой спастико-гипокинетической дизартрии.
4) дистония и болезненные мышечные спазмы (чаще мимическую мускулатуру и стопы- при ходьбе тыльне сгибание и ротация стоп).
5) гипомимия, замедленность движений, блефароспазм.
6) умеренные признаки пирамидной и мозжечковой недостаточности.
7) в анамнезе интоксикация (марганцевая и др.).
1) наличие постурально-кинетического тремора;
2) часто двусторонний дебют заболевания;
3) вовлечение в процесс дрожательного компонента головы и голосовых связок;
4) отсутствие выраженной гипокинезии;
5) отсутствие эффекта дофаминзаместительной терапии;
6) длительное доброкачественное течение.
Деменция с тельцами Леви является более диффузным дегенеративным процессом, чем болезнь Паркинсона, т.к. клеточные включения, помимо черной субстанции встречаются в коре головного мозга (преимущественно в височных долях) и базальном ядре Мейнерта. Диагностика деменции с тельцами Леви зачастую затруднена, учитывая клиническое и морфологическое сходство с болезнью Паркинсона. Оба этих заболевания характеризуются паркинсонизмом и деменцией, но в отличие от болезни Паркинсона признаки деменции превалируют как по клинической картине, так и во временном аспекте (возникают в течение года). Кроме того, в ряде случаев имеются характерные клинические особенности:
1) зрительные галлюцинации, иногда с другими психическими расстройствами (бред преследования и др.);
2) ранняя постуральная неустойчивость с частыми падениями;
3) выраженные нарушения внимания и психической активности с преходящими эпизодами спутанности сознания;
4) раннее развитие депрессии.
Прогрессирующий надъядерный паралич (болезнь Стила-Ричардсона – Ольшевского) относится к мультисистемным дегенерациям с вовлечением в процесс базальных ганглиев, стволовых ядер, четверохолмия, зубчатых ядер мозжечка, кору лобных долей. Встречается в 2-5% случаев. В его основе лежит агрегация тау-протеина с образованием внутриклеточных включений, в связи с чем заболевание относят к таупатиям. Наиболее характерными признаками являются:
1) паралич вертикального взора, чаще вниз;
2) ранняя постуральная неустойчивость;
3) развитие деменции, преимущественно лобного типа;
4) раннее развитие псевдобульбарного синдрома;
6) быстропрогрессирующее течение (3-5 лет);
7) симметричность симптоматики с редко встречающимся тремором.
Мультисистемная атрофия (МСА)– спорадическая мультисистемная дегенерация, поражающая базальные ганглии, оливы, мост, мозжечок, боковые рога спинного мозга. Встречается в 2-6% случаев. МСА, также как болезнь Паркинсона и деменция с тельцами Леви, относится к синуклеинопатиям, но накопление альфа-синуклеина присходит преимущественно в олигодендроцитах. Отличительные особенности:
1) наличие паркинсоновского синдрома с нехарактерным для болезни Паркинсона постурально-кинетическим тремором;
2) раннее развитие тяжелой вегетативной патологии, включая ортостатическую гипотензию;
3) быстрое прогрессирование с ранним развитием постуральной неустойчивости и псевдобульбарного синдрома;
4) наличие мозжечковой и пирамидной симптоматики в половине случаев при отсутствии деменции и других психических нарушений;
5) на МРТ признаки атрофии моста и мозжечка, снижение интенсивности сигнала от скорлупы.
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) – наследственное дисметаболическое заболевание нервной системы, связанное с накоплением меди в головном мозге, печени и роговице. Характерные диагностические признаки:
1) наличие пигментного роговичного кольца Кайзера-Флейшера;
2) снижение содержания связанного с медью церулоплазмина в крови;
4) Наличие акинетико-ригидного синдрома, дистонии, постурального тремора и миоклоний.
В этой главе отражена картина не всех нозологических форм, сопровождающихся синдромом паркинсонизма. Внедрение в медицинскую практику современных методов обследования, особенно нейровизуализации, позволяет диагностировать такие заболевания, как болезнь Альцгеймера, деменция с тельцами Леви, ВИЧ-инфекции и др. Учитывая многообразие нозологических форм паркинсонизма, наиболее значимым звеном в диагностике болезни Паркинсона являются четко обозначенные диагностические критерии, применяемые в мировой практике.
*Импакт фактор за 2018 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Читайте в новом номере
ММА имени И.М. Сеченова
Клиническая картина

Болезнь Паркинсона. На рисунке А показан участок мозга здорового человека с хорошо пигментированным черным веществом. На рисунке В - участок мозга пациента, страдающего болезнью Паркинсона. Заметно отсутствие пигментации черного вещества.
П – это синдром, который может встречаться в рамках разных заболеваний. Наиболее часто причиной П является идиопатический паркинсонизм, или болезнь Паркинсона (БП). П часто наблюдается в рамках других идиопатических дегенеративных заболеваний нервной системы. Последние часто называют паркинсонизмом–плюс. К этой группе относятся мультисистемная атрофия, прогрессирующий надъядерный паралич, болезнь диффузных телец Леви, кортико-базальная дегенерация. Нередко встречается симптоматический П (не связанный с первичным дегенеративным заболеванием нервной системы). К нему относят лекарственный, сосудистый, посттравматический, постэнцефалитический, токсический П, П при опухолях головного мозга и гидроцефалии. П характеризует также клиническую картину некоторых наследственных дегенеративных заболеваний ЦНС. К их числу относятся гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова), спиноцеребеллярные атаксии, семейная кальцификация базальных ганглиев, болезнь Гентингтона и др. Таким образом, синдром П не является синонимом БП.
Диагноз и дифференциальный диагноз
Эссенциальный тремор
На первом этапе диагностической работы необходимо убедиться, что у больного действительно имеется синдром П. Наиболее часто за истинный П ошибочно принимают эссенциальный тремор (ЭТ) и своеобразные изменения походки, связанные с сосудистой патологией головного мозга. Таким пациентам очень часто неправильно ставят диагноз БП.
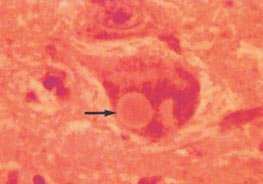
Тельца Леви при болезни Паркинсона. В цитоплазме нейрона определяется эозинофильное ядро окруженное неокрашенной зоной.
Последние, как правило, выявляют множественные мелкие очаги сосудистого происхождения (лакуны) в области базальных ганглиев и/или изменения белого вещества полушарий головного мозга (лейкоареоз). Следует иметь в виду, что схожая клиническая картина может возникать иногда при нормотензивной гидроцефалии и реже – при опухолевом поражении головного мозга. При нормотензивной гидроцефалии наряду с нарушением ходьбы по типу описанного выше имеют место деменция и расстройство функции тазовых органов (эту триаду симптомов называют триадой Хакима–Адамса). МРТ головы, как правило, обнаруживает резко расширенные боковые желудочки.
Болезнь Паркинсона
Диагноз БП – это клинический диагноз. Параклинические методы исследования обычно выявляют неспецифические изменения. Нередко у больных с классической БП в головном мозге имеются сосудистые изменения. Это, однако, ни в коем случае не является свидетельством сосудистого генеза болезни. Просто у большинства пожилых людей, в том числе практически здоровых, подобные изменения могут иметь место. Тем не менее, в некоторых случаях на БП наслаивается атеросклеротический псевдопаркинсонизм, что может быть причиной необычной для БП походки и ранних постуральных нарушений.
Лекарственный паркинсонизм
Лекарственный П может быть обусловлен препаратами, которые воздействуют на пресинаптические дофаминовые нейроны черной субстанции, истощая запасы дофамина в них (например, резерпин) или, наиболее часто, нейролептиками, которые блокируют постсинаптические дофаминовые рецепторы, такими как производные фенотиазина (хлорпромазин), бутирофеноны (галоперидол), тиоксантины (флупентиксол) и бензамиды (сульпирид). Эти препараты часто применяются при психических заболеваниях. Также П может быть вызван прохлорперазином (используется при рвоте, головокружении и неустойчивости), метоклопрамидом (применяется при заболеваниях желудочно-кишечного тракта, для купирования тошноты и рвоты). П может быть обусловлен также циннаризином, который является атипичным блокатором кальциевых каналов (применяется при вестибулярных расстройствах). Комбинация нейролептиков и антидепрессантов также может быть причиной П.
В психиатрических лечебницах лекарственный П встречается часто. Гипомимия и ахейрокинез – настолько обычное явление среди этого контингента больных, что психиатры часто даже не обращают на них внимание. Тремор встречается менее часто, но может иметь вид классического паркинсонического дрожания. Более того, лекарственный П может быть асимметричным, подобно БП, и нередко совершенно не отличим от БП. Одним из признаков, по которому следует заподозрить лекарственный П, является наличие наряду с акинетико-ригидным синдромом насильственных движений в виде, например, оро-мандибулярной дискинезии (непроизвольные жевательные и/или сосательные движения) или дистонических явлений (спастической кривошеи, окулогирных кризов), стереотипий, акатизии (неусидчивость). Нередко тяжелый лекарственный П сопровождается выраженной дизартрией и дисфагией. Индивидуальная чувствительность к дофаминблокирующим препаратам очень разнообразна. Некоторые пациенты без каких-либо проблем переносят длительное лечение большими дозами этих веществ, тогда как у других побочные явления развиваются уже на малых дозах. Чаще, однако, признаки лекарственного П возникают при приеме больших доз нейролептиков. Лекарственный П обычно развивается постепенно в течение дней или недель. У большинства больных первые признаки появляются через 3 нед после начала лечения. Наиболее часто встречающиеся дебютные признаки – гипомимия и недостаточное раскачивание рук во время ходьбы.
Течение лекарственного П может быть различным. В большинстве случаев он постепенно, в течение нескольких недель, а иногда и дней, проходит после прекращения приема вызвавшего его препарата. Тем не менее нередки случаи, когда П длится в течение месяцев, иногда почти год. Такая ситуация наблюдается при применении нейролептических препаратов, способных к депонированию. В редких случаях лекарственный П не проходит и продолжает прогрессировать, несмотря на прекращение приема вызвавшего его агента. Подобные случаи чаще встречаются среди пожилых людей. Считается, что в таких случаях прогрессирует не сам по себе лекарственный П, а начинает развиваться БП.
Мультисистемная атрофия
Мультисистемная атрофия (МСА) – это спорадическое заболевание, возникающее у взрослых лиц, при котором в отличие от БП дегенерации подвергается не только нигро-стриарная система, но также множество других образований ЦНС, включая мозжечок и его связи, пирамидные пути и образования вегетативной нервной системы (отсюда и происходит название болезни). Соответственно клинически МСА характеризуется сочетанием П, мозжечковых нарушений, пирамидных расстройств и прогрессирующей вегетативной недостаточности (ПВН). П при МСА обусловлен не только поражением клеток черной субстанции, что вызывает дефицит дофамина, но также дегенерацией тех постсинаптических рецепторов, с которыми должен взаимодействовать дофамин.
В клинической картине МСА может преобладать та или иная симптоматика. Те случаи, при которых на первый план выступает П, обозначают термином нигро-стриальная дегенерация (СНД); если в клинической картине ведущим является мозжечковый синдром, это состояние называют оливо-понто-церебеллярной атрофией (ОПЦА); случаи, когда ядром клинической картины является ПВН, обозначают эпонимическим названием – синдром Шая–Дрейджера (СШД).
Несмотря на все различия, нередки ситуации, когда МСА невозможно отличить от БП. В таких случаях следует в основном ориентироваться на эффект препаратов леводопы. При БП эти препараты оказывают драматический положительный эффект, тогда как при МСА этот эффект не столь выражен, кратковременен, а нередко отсутствует совсем. Это обусловлено поражением постсинаптических рецепторов, с которыми должна взаимодействовать леводопа.
Признаки, при наличии которых у больных с П следует подумать об МСА, следующие: быстрое прогрессирование симптоматики; рано развившиеся нарушения равновесия и падения; отсутствие улучшения при лечении препаратами леводопы или недостаточный эффект этих препаратов; ПВН; пирамидные и/или мозжечковые знаки; похолодание, зябкость конечностей; контрактуры; диспропорциональный антеколлис; выраженная дисфония, дисфагия или дизартрия; респираторный стридор; нерегулярный тремор или миоклонии.
Прогрессирующий надъядерный паралич
Болезнь диффузных телец Леви
В последнее время стали выделять новую нозологическую форму, протекающую с синдромом П – болезнь диффузных телец Леви (БДТЛ). Тельца Леви – это внутриклеточные эозинофильные цитоплазматические включения, которые обнаруживаются в клетках черного вещества при БП и считаются маркером этой болезни. При БДТЛ они встречаются не только в черном веществе, но в большом количестве широко диссеминированы по всему головному мозгу. Диагноз БДТЛ – патоморфологический диагноз. Клинически это заболевание характеризуется П, который обычно хорошо лечится препаратами леводопы, наряду с деменцией с выраженными зрительными галлюцинациями. Типичным является также флуктуация выраженности расстройств высших психических функций – в основном за счет изменения способности концентрации внимания.
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) и другие заболевания
Существует несколько более редких причин истинного или псевдопаркинсонизма. Одна из них, о которой следует всегда вспомнить при наличии П у людей моложе 45 лет (в том числе у детей), это гепатолентикулярная дегенерация, или болезнь Вильсона-Коновалова. Это наследственное заболевание, при котором отмечается нарушение метаболизма меди в организме из-за недостаточности фермента церулоплазмина. В результате медь в избыточном количестве откладывается в печени, базальных ганглиях и вокруг радужной оболочки глаза. Болезнь Вильсона-Коновалова следует подозревать не только при наличии П у молодых людей, но и при возникновении у них других признаков поражения экстрапирамидной системы (например, дистонии) или психических расстройств. Диагностика основана на обнаружении с помощью щелевой лампы отложения меди вокруг радужки – кольцо Кайзера–Флейшера. Последнее на стадии неврологических проявлений имеет место у 98% больных. Диагностическое значение имеет также исследование экскреции меди с мочой и концентрации церулоплазмина в крови. Последнее, однако, в отсутствие кольца Кайзера–Флейшера и нормальной экскреции меди не имеет диагностической значимости. Если ситуация продолжает оставаться неясной, проводится биопсия печени или генетическое тестирование. Болезнь Вильсона-Коновалова довольно успешно лечится с помощью D-пеницилламина и препаратов цинка в сочетании с диетой.
Токсический П может иметь место при интоксикации марганцем. Нередко встречается П, вызванный токсином МФТП (1-метил-4-фенил- 1, 2, 3, 6-тетрагидропиридин), так как оно избирательно поражает черную субстанцию, вызывая гибель дофаминовых нейронов. Схожий с этим веществом токсин вырабатывается при кустарном приготовлении некоторых наркотических веществ, поэтому П нередко наблюдается среди молодых людей, страдающих наркоманией. Наряду с П при этом могут отмечаться пирамидные и псевдобульбарные симптомы.
П встречается при энцефалопатии боксеров. Считается, что в таком случае этиологическую роль играют регулярно получаемые множественные черепно-мозговые травмы. Следует помнить, что энцефалопатия боксеров – практически единственная ситуация, когда черепно-мозговые травмы играют роль в развитии П. В целом у людей, не занимающихся боксом, наличию черепно-мозговой трамы в анамнезе (одной или нескольких) не придается этиологическое значение.
П иногда может иметь место при хорее Гентингтона, которое обычно протекает с выраженными гиперкинезами. Это наследственное заболевание обычно дебютирует на четвертом десятилетии жизни, однако встречаются случаи и более раннего начала. Обычно с акинетико-ригидным синдромом протекает так называемая ювенильная форма хореи Гентингтона (форма Вестфаля). Встречаются также семейные случаи П, в том числе ювенильного. Поэтому у всех больных, имеющих синдром П, сбор наследственного анамнеза имеет большое значение.
Акинезия и ригидность могут иногда наблюдаться при болезни Альцгеймера, Пика, при которых ядром клинической картины является прогрессирующая деменция. При болезни Альцгеймера в первую очередь страдает память, а при болезни Пика на первый план выступает распад личности. При этих заболеваниях П чаще проявляется умеренной акинезией, ригидность выражена меньше, а паркинсонический тремор практически никогда не наблюдается.
Постэнцефалитический П в наше время практически не встречается. Много случаев наблюдалось после пандемии летаргического энцефалита в начале XX столетия. С тех пор в литературе описано только несколько случаев.
Особая эндемическая форма П наблюдается на острове Гуам и в некоторых других местах восточной части Тихого океана. В этих местах отмечается много случаев П в сочетании с амиотрофиями и деменцией (гуамский комплекс паркинсонизм – деменция). Патоморфологически мозг этих больных выглядит, как мозг больных ПНП. Одинаковыми являются также гистологические маркеры.
Читайте также: