
Врожденные аномалии черепных нервов
Нейросенсорная глухота. Клиника.Диагностика.
Врожденные пороки развития обонятельного нерва изолированно встречаются редко; отсутствие обонятельных трактов обычно сочетается с лизэнцефалией.
Пороки развития зрительного нерва могут проявляться атрофией его волокон с разрастанием глии, аплазией или гипоплазией диска, его избыточной миелинизацией и колобомами.
Пороки развития глазодвигательных нервов чаще бывают односторонними и связаны с аплазией ядер или нарушением строения глазницы. При недоразвитии заднего продольного пучка может быть парез взора в сторону или врожденный крупноразмашистый нистагм, сочетающийся с ритмичными движениями головы в сторону.
Аплазия ядер тройничного нерва наблюдается крайне редко. Сохранение эмбриональной связи между двигательными ядрами тройничного нерва и ядрами глазодвигательного нерва обусловливает сочетанные движения глаз и нижней челюсти,
Врожденные поражения лицевого нерва могут быть связаны с аплазией ядер, атрезией костного канала или шилососцевидного отверстия.
Синдром Мебиуса — врожденная аплазия ядер отводящего и лицевого нервов (нарушение функции).
Лицо ребенка маскообразное, без морщин, с открытым ртом и незакрывающимися глазами. Углы рта опущены. Глазные яблоки не отводятся в стороны. Иногда наблюдается полная наружная офтальмоплегия вследствие аплазии ядер глазодвигательных нервов или полного отсутствия наружных мышц глаза.
Менее постоянно при синдроме Мебиуса отмечается недоразвитие ядер V, VIII, IX, X, XII черепных нервов. Поражение каудальной группы ядер проявляется клиникой бульварного паралича и быстро приводит к летальному исходу.
Кроме аплазии ядер, могут наблюдаться другие аномалии развития: микрофтальм, деформации ушей, аномалии прикуса, лишние пальцы и отсутствие пальцев, синдактилия, брахидактилия, косолапость, недоразвитие грудных мышц. Интеллект снижен примерно у 40% больных. Синдром Мебиуса следует дифференцировать от травматических поражений лицевого нерва в родах.
92. Эпилепсия. Этиология. Международная классификация.
Эпилепсия – хроническое заболевание ГМ, проявляющееся повторными, непровоцирующими приступами с нар. двиг., чувств., вегетатив., психич ф-ций, обусловленных чрезмерными нейрональными разрядами в сером в-ве коры ГМ.
Этиология. Спонтанная генерализованная или очаговая гиперсинхронная зарядка-разрядка нейронов коры ГМ из-за возникновения на кл. мембране пароксизмального деполяризационного сдвига. При этом возник пролонгация деполяризации.
Причина ПДС - нарушение баланса возб. и торм. медиаторов.
Локализация - локально обусловленные (фокальные, локальные, парциальные)
Формы - генерализованные и имеющие черты парциальных и генерал
Этиология - симптоматические, криптогенные, идиопатические
Возраст дебюта - новор, млад., детские, юношеские, взрослые
Виды приступов - абсансы, миоклонич абсансы
Что такое синдром Мебиуса?
Синдром Мебиуса (врожденная лицевая диплегия) — это редкое неврологическое заболевание, характеризующееся слабостью или параличом множественных черепных нервов, чаще всего 6-го (отводящего) и 7-го (лицевого) нерва. Иногда поражаются другие черепные нервы. Расстройство присутствует при рождении (врожденное).
Если задействован 7-й нерв, человек с синдромом Мебиуса не может улыбаться, хмуриться, сморщивать губы, поднимать брови или закрывать веки. Если поражен 6-й нерв, глаз не способен повернуться наружу за среднюю линию. Другие аномалии включают недоразвитие грудных мышц и дефекты конечностей.
Синдром Мебиуса не прогрессирует. Точная причина неизвестна. Похоже, что синдром возникает случайно (спорадически) в большинстве случаев; однако, некоторые случаи происходят в семьях, предполагая, что может быть генетический компонент.
Признаки и симптомы
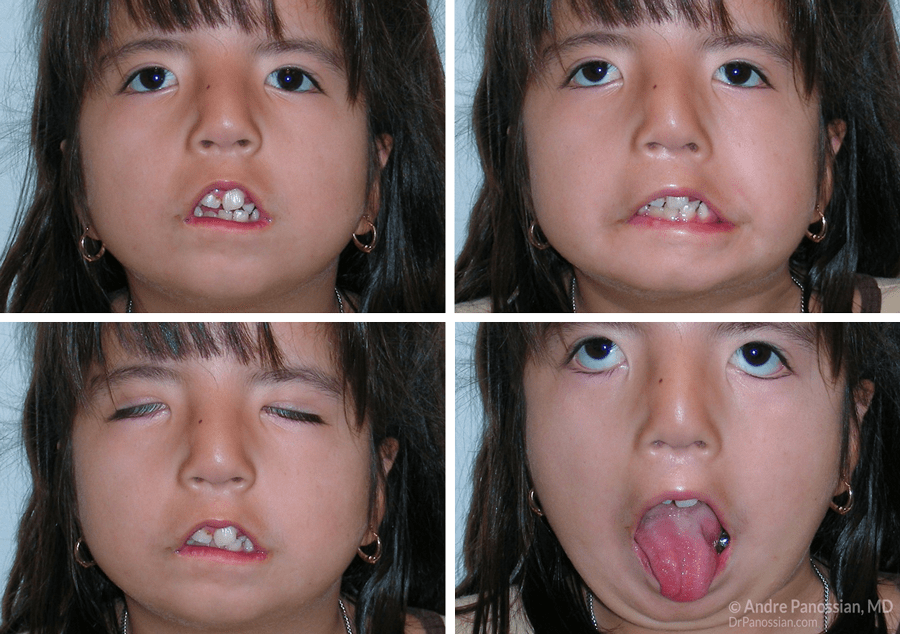
Нарушения и тяжесть синдрома Мебиуса сильно различаются у разных людей. Классически принятые диагностические критерии включают:
- паралич лица или слабость, затрагивающие, по крайней мере, одну, но обычно обе стороны лица (7-й черепной нерв);
- паралич бокового движения глаз (6-й черепной нерв);
- сохранение вертикальных движений глаз.
Реже могут поражаться другие черепные нервы, в том числе 5-й, 8-й, 9-й, 10-й, 11-й и 12-й.

У пострадавших детей также могут возникнуть проблемы с кормлением, в том числе проблемы с глотанием и плохое сосание. Также, язва роговицы может возникнуть, потому что веки остаются открытыми во время сна.
Существует множество дополнительных отклонений. У некоторых детей с синдромом Мебиуса короткий, неправильно сформированный язык и/или аномально маленькая челюсть (микрогнатия). Также может присутствовать волчья пасть. Эти нарушения способствуют затруднению питания и дыхания. Дети с расщелиной неба склонны к ушным инфекциям (средний отит). Могут быть аномалии наружного уха, включая недоразвитие наружной части уха (микротия) или полное отсутствие наружной части уха (анотия). Если поражен 8-й черепной нерв, существует вероятность потери слуха. Зубные аномалии не редкость. У некоторых пострадавших детей возникают проблемы с речью и задержки в развитии речи.
Пороки развития скелета конечностей встречаются у более половины детей с синдромом Мебиуса. Пороки развития нижних конечностей включают косолапость и недоразвитие голеней; на верхних конечностях могут обнаружится перепонки пальцев (синдактилия), недоразвитие или отсутствие пальцев и/или недоразвитие рук. У некоторых детей может быть патологическое искривление позвоночника (сколиоз), а примерно у 15% пациентов также наблюдается недоразвитие грудных мышц и груди на одной стороне тела.
У некоторых затронутых синдромом детей наблюдается задержка в достижении определенных вех, таких как ползание или ходьба, скорее всего, из-за слабости верхней части тела; однако большинство детей в конечном итоге наверстывают упущенное. Синдром Мебиуса редко связан с незначительной умственной отсталостью. Некоторые дети были классифицированы как имеющие расстройство аутистического спектра. Точная связь между синдромом Мебиуса и аутизмом неизвестна.
Некоторые исследования показали, что расстройства аутистического спектра чаще встречаются у детей с синдромом Мебиуса; другие исследования не подтвердили это и предполагают, что любые такие отношения завышены.
Синдром Мебиуса часто связан с различными социальными и психологическими последствиями. Отсутствие мимики и неспособность улыбаться могут заставить наблюдателей неверно истолковать, что думает или чувствует больной человек. Хотя клиническая тревога и депрессия не часто встречаются у детей и подростков с синдромом Мебиуса, пострадавшие могут избегать социальных ситуаций из-за опасений и разочарования.
Причины
Большинство случаев синдрома Мебиуса возникают случайно по неизвестным врачам причинам (спорадически) при отсутствии семейного анамнеза расстройства. Спорадические мутации в генах PLXND1 и REV3L также были идентифицированы у ряда пациентов, и было подтверждено, что они вызывают совокупность результатов, согласующихся с синдромом Мебиуса, при введении их животным.
Также есть редкие случаи, зарегистрированные в семьях. Скорее всего, синдром Мебиуса является многофакторной патологией, что означает, что генетические факторы и факторы окружающей среды играют определенную причинную роль. Возможно, что в разных случаях существуют разные причины (гетерогенность).
В семейных случаях есть свидетельства того, что синдром Мебиуса наследуется как аутосомно-доминантный признак. Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения гена) у пострадавшего человека. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% для каждой беременности, независимо от пола ребенка.
Спектр находок при синдроме Мёбиуса свидетельствует о пороке развития заднего мозга. Для объяснения причины синдрома Мебиуса было предложено несколько разных теорий. Одна из гипотез заключается в том, что расстройство является результатом ослабленного или прерванного кровотока (ишемии) к развивающемуся плоду во время беременности (внутриутробно). Недавние исследования показывают, что недостаток крови влияет на определенные области нижнего ствола мозга, которые содержат ядра черепных нервов. Это отсутствие кровотока может быть вызвано экологической, механической или генетической причиной. Тем не менее, причина синдрома остается неясной, и необходимы более фундаментальные и клинические исследования.
Затронутые группы населения
Синдром Мебиуса поражает мужчин и женщин в равных количествах. Расстройство присутствует при рождении. Точная частота и распространенность синдрома Мебиуса неизвестны. Согласно одной из оценок, заболеваемость в США составляет 1 случай на 50 000 живорождений.
Схожие расстройства
Симптомы и признаки следующих расстройств могут быть схожими с симптомами синдрома Мебиуса. Сравнения могут быть полезны для дифференциальной диагностики.
В медицинской литературе ведутся споры о том, представляют ли синдром Мебиуса, синдром Польши и синдром Польши-Мебиуса три различных расстройства или три различных проявления одного расстройства.
Наследственный врожденный паралич лица характеризуется изолированным параличом лицевого нерва, и подгруппы болезни имеют другие связанные признаки, включая потерю слуха и косоглазие. Наследственный врожденный паралич лица отличается от синдрома Мебиуса наличием полного горизонтального взгляда. У пациентов с данной патологией были идентифицированы мутации HOXB1 на хромосоме 17q21.
Существуют и другие причины врожденного паралича лица, в частности, микросомия полушария лица и, возможно, родовая травма. Есть также приобретенные типы лицевого паралича. Синдром Мелькерссона-Розенталя характеризуется внезапным началом в детстве/подростковом возрасте, двусторонней или односторонней слабостью лица, хроническим отеком лица (особенно губ) и нахмуренным языком.
Другие возможные причины:
- инфекции, такие как синдром Рамсей-Ханта;
- системные и неврологические расстройства, включая детский церебральный паралич, врожденная мышечная дистрофия, спинальная мышечная атрофия, синдром Гийена-Барре, рассеянный склероз и аутоиммунные расстройства;
- травма головного мозга;
- опухоли или повреждения ствола мозга.
Диагностика
Диагноз синдрома Мебиуса основан на характерных признаках/симптомах, подробном анамнезе пациента и тщательной клинической оценке. Не существует диагностических тестов, подтверждающих диагноз синдрома Мебиуса. Чтобы исключить другие причины лицевого паралича могут быть выполнены дополнительные специальные тесты.
Лечение
Специального курса терапии синдрома Мебиуса не существует. Лечение синдрома Мебиуса направлено на терапию конкретного нарушения у каждого человека. Обычно этими детьми занимается многопрофильная команда, часто в черепно-лицевом центре. К вовлеченным специалистам относятся:
- педиатры;
- неврологи;
- пластические хирурги;
- специалисты по уху, носу и горлу (отоларингологи);
- ортопеды;
- зубные специалисты;
- логопеды;
- специалисты по оценке и лечению нарушений слуха (аудиологи);
- специалисты по лечению нарушений зрения (офтальмологи) и другие медицинские работники.
Самая последняя процедура включает микрососудистый перенос мышцы бедра к лицу и соединение нервов, которые обычно снабжают мышцу жевательной мышцы. Эта операция показала замечательные результаты с точки зрения речи, мобильности лица и самооценки.
Физическая терапия может быть необходима для людей с различными ортопедическими нарушениями. Трудовая терапия также может быть полезной, особенно у пациентов с аномалиями рук, пальцев рук и ног. Логопедия также может быть необходима для некоторых пострадавших детей. Косоглазие обычно корректируется хирургическим путем, хотя некоторые врачи рекомендуют отложить эти процедуры, поскольку состояние иногда улучшается с возрастом. Операции также могут быть необходимы для различных пороков развития скелета, затрагивающих конечности и челюсти.
Прогноз
Лекарств от синдрома Мебиуса нет. Несмотря на нарушения, которые характеризуют расстройство, надлежащий уход и лечение синдрома дают многим людям нормальную продолжительность жизни.
Врожденные пороки развития обонятельного нерва изолированно встречаются редко; отсутствие обонятельных трактов обычно сочетается с лизэнцефалией.
Пороки развития зрительного нерва могут проявляться атрофией его волокон с разрастанием глии, аплазией или гипоплазией диска, его избыточной миелинизацией и колобомами.
Пороки развития глазодвигательных нервов чаще бывают односторонними и связаны с аплазией ядер или нарушением строения глазницы. При недоразвитии заднего продольного пучка может быть парез взора в сторону или врожденный крупноразмашистый нистагм, сочетающийся с ритмичными движениями головы в сторону.
Аплазия ядер тройничного нерва наблюдается крайне редко. Сохранение эмбриональной связи между двигательными ядрами тройничного нерва и ядрами глазодвигательного нерва обусловливает сочетанные движения глаз и нижней челюсти,
Врожденные поражения лицевого нерва могут быть связаны с аплазией ядер, атрезией костного канала или шилососцевид-ного отверстия.
Синдром Мебиуса — врожденная аплазия ядер отводящего и лицевого нервов аут-дом.
Лицо ребенка маскообразное, без морщин, с открытым ртом и незакрывающимися глазами. Углы рта опущены. Глазные яблоки не отводятся в стороны. Иногда наблюдается полная наружная офтальмоплегия вследствие аплазии ядер глазодвигательных нервов или полного отсутствия наружных мышц глаза.
Менее постоянно при синдроме Мебиуса отмечается недоразвитие ядер V, VIII, IX, X, XII черепных нервов. Поражение каудальной группы ядер проявляется клиникой бульварного паралича и быстро приводит к летальному исходу.
Кроме аплазии ядер, могут наблюдаться другие аномалии развития: микрофтальм, деформации ушей, аномалии прикуса, лишние пальцы и отсутствие пальцев, синдактилия, брахидактилия, косолапость, недоразвитие грудных мышц. Интеллект снижен примерно у 40% больных. Синдром Мебиуса следует дифференцировать от травматических поражений лицевого нерва в родах.
Эпилепсия. Этилогия. Международная классификация.
Эпилепсия- хронич. Заб-ие ГМ, проявл., повторными, непровоцирующими приступами с нар. Двиг., чувств., вегетатив.,психич ф-ций обусловленных черезмерными нейрональными разрядами в сером в-ве коры ГМ.
Этиология. Спонтанная генерализованная или очаговая гиперсинхронная зарядка-разрядка нейронов коры ГМ из-за возникновения на Кл.мембране пароксизмального деполяризационного сдвига. При этом возник пролонгация деполяризации.
Причина ПДС- нар баланса возб и торм медиаторов
Патогенез. Нар в рец К-Na каналов или избыточный выброс глутамата и аспартата-предшеств тормоз мед-ра ГАМК.
Локализация- локально обусловленные (фокальные, локальные, парциальные)
Формы- генерализованные и имеющие черты парциальных и генерал
Этиология- симптоматические, криптогенные, идиопатические
Возраст дебюта- новор, млад., детские, юношеские, взрослые
Виды приступов- абсансы, миоклонич абсансы
Международная классификация эпилептических припадков.
Припадок- первичное пароксизмальное расс-во функций ГМ
Генерализованные приступы ( обе гемисферы синхр вовлечены)
- типичные абссансы- кортико-таламич, замирание с выкл созн
- эпилептич спазмы- с доп сенсорно-мот зоны, билатеральные сокр., созн вариабельно
- генерализ эпилептич миоклонус
- миатоническиекотико-талам-ретик, каскад приседаний и падений, созн сохр
Фокальные приступы (1 гемисфера или асинхр 2)
- сенсорные (с прост. И слож. Симптомами)
- моторные (клонич- лоб.доля, контрлат- клонич судороги, сознание сохр./., тонич.- медиал часть поля 6, 2-х стор судороги в конеч, созн сохр, с тиич. Автоматизмами, негатив миоклонус, ингибиторные- негатив зона в нижЛизв, нев-ть выпол движ в отд конеч)
- геластические (смеха)- ниж Л.И. расп на гипот, приступы смеха, созн сохр.
- гемиклонические- первич мотор зона ЛД, клонич судор лица, руки и ноги на 1 стор, созн сохр
- вторич генерализованные- первич мотор зона ЛД, фокальный приступ, созн утрачено
Так же атонич., атипич абсансы, эписпазмы
Дата добавления: 2020-01-07 ; просмотров: 39 ;
Аномалии развития черепных нервов проявляются аплазией их ядер, атрофией волокон или их сочетанием. Часто наряду с недоразвитием волокон черепных нервов отмечается нарушение развития костных каналов, через которые они проходят.
Рис. 86. Синдром Мебиуса у детей с множественными пороками развития.
Врожденные пороки развития обонятельного нерва изолированно встречаются редко; отсутствие обонятельных трактов обычно сочетается с лизэнцефалией.
Пороки развития зрительного нерва могут проявляться атрофией его волокон с разрастанием глии на всем протяжении нерва, аплазией или гипоплазией диска, его избыточной миелинизацией и колобомами. Эти пороки бывают односторонними или двусторонними, изолированными или сочетаются с аномалиями развития мозга и глаз. Зрение резко снижено или отсутствует с рождения.
Пороки развития глазодвигательных нервов чаще бывают односторонними и связаны с аплазией ядер или нарушением строения глазницы. Они проявляются сходящимся, расходящимся косоглазием, нарушением зрачковых реакций, птозом. Изолированный врожденный птоз наблюдается при аплазии боковых парных ядер глазодвигательного нерва. При недоразвитии заднего продольного пучка может быть парез взора в сторону или врожденный крупноразмашистый нистагм, сочетающийся с ритмичными движениями головы в сторону.
Аплазия ядер тройничного нерва наблюдается крайне редко. Сохранение эмбриональной связи между двигательными ядрами тройничного нерва и ядрами глазодвигательного нерва обусловливает сочетанные движения глаз и нижней челюсти, известные под названием синкинезии Маркуса—Гунна. Сийкинезия Маркуса—Гунна известна в трех вариантах: 1) непроизвольное поднимание птозированного века при открывании или закрывании рта; 2) поднимание птозированного века при движении челюсти вбок; 3) ассоциация движений век с движениями нижней челюсти.
Врожденные поражения лицевого нерва могут быть связаны с аплазией ядер, атрезией костного канала или шилососцевидного отверстия.
Синдром Мебиуса — врожденная аплазия ядер отводящего и лицевого нервов (рис. 86). Многие случаи этого синдрома спорадические, но имеются описания семей с аутосомно-рецессивным и аутосомно-доминантным наследованием.
Частота синдрома. Мебиуса составляет 1 случай на 20 000 новорожденных.
Лицо ребенка маскообразное, без морщин, с открытым ртом и незакрывающимися глазами. Углы рта опущены. Глазные яблоки не отводятся в стороны. Иногда наблюдается полная наружная офтальмоплегия вследствие аплазии ядер глазодвигательных нервов или полного отсутствия наружных мышц глаза.
Менее постоянно при синдроме Мебиуса отмечается недоразвитие ядер V, VIII, IX, X, XII черепных нервов. Поражение каудальной группы ядер проявляется клиникой бульбарного паралича и быстро приводит к летальному исходу. Пороки развития каудальной группы черепных нервов изолированно встречаются редко, а в сочетании с другими нарушениями обнаруживаются при пороках развития мозгового ствола, синдромах платибазии, базилярной инвагинации, Клиппеля—Фейля и др.
Кроме аплазии ядер, могут наблюдаться другие аномалии развития: микрофтальм, деформации ушей, аномалии прикуса, лишние пальцы и отсутствие пальцев, синдактилия, брахидактилия, косолапость, недоразвитие грудных мышц. Интеллект снижен примерно у 40% больных. Синдром Мебиуса следует дифференцировать от травматических поражений лицевого нерва в родах.
Дата добавления: 2015-02-06 | Просмотры: 1275 | Нарушение авторских прав
Анэнцефалия — полное отсутствие мозговых гемисфер, сочетающееся с дефектом костей черепа. Кости мозгового черепа полностью отсутствуют или представлены рудиментарными хрящевыми образованиями, иногда островками лобных и теменных долей. Стволовые образования мозга также недоразвиты. Частота анэнцефалии составляет 5—8 случаев на 10 000 новорожденных; в 70—80% случаев отмечаются мертворождения. Продолжительность жизни детей, родившихся живыми, составляет не более 10 дней, что связано с тяжелым нарушением жизненно важных функций.
Гидранэнцефалия — врожденный порок развития мозга, при котором большие полушария полностью отсутствуют и замещены полостью, заполненной жидкостью. Иногда имеются рудименты лобных и затылочных отделов коры. Мозговой ствол и базальные ганглии сформированы в достаточной мере. Частота гидранэнцефалии составляет 1 случай на 18 000 новорожденных.
У новорожденных с таким дефектом отсутствуют кости черепа. Пузырь с жидкостью, замещающий большой мозг, покрыт только мягкими тканями. Врожденные рефлекторные реакции у детей сохранены в большей или меньшей степени. С рождения отмечаются частые судороги. С первых месяцев жизни выявляются грубые моторные и психические нарушения. Обычно дети умирают в первые 3—4 мес жизни.
При трансиллюминации черепа выявляется расширенная зона свечения в области мозговых гемисфер, при пневмоэнцефалографии — значительное расширение желудочков мозга, которые иногда занимают всю полость черепа. Ангиография указывает на отсутствие мозговых сосудов в области полушарий.
В основе профилактики анэнцефалии и гидранэнцефалии лежит антенатальная диагностика этих состояний, которая обычно проводится в тех случаях, если в семье был ребенок с подобного рода нарушением.
Антенатальный диагноз ставится на основании многоводия, при котором количество околоплодной жидкости превышает норму в 8— 10 раз, а также уменьшения количества эстрогенов в моче беременной женщины, увеличения содержания альфафетопротеина в амниотической жидкости. Диагностике помогают амниофетоскопия и рентгенологическое исследование плода.
ГОЛОПРОЗЭНЦЕФАЛИЯ
Голопрозэнцефалия — порок развития мозга, при котором мозговые гемисферы не разделены на полушария, а мозговые желудочки представлены одной полостью. Этот дефект образуется на стадии разделения мозговых гемисфер. Он может сочетаться с отсутствием обонятельных луковиц и обонятельного тракта, расщеплением губы, недоразвитием глаз, циклопией. Голопрозэнцефалия может быть проявлением трисомии 13—15 хромосом. Частота этого порока составляет 1 случай на 15 000 новорожденных.
ЛИЗЭНЦЕФАЛИЯ
Лизэнцефалия — недоразвитие мозговых извилин, при этом поверхность мозговых гемисфер гладкая. В основе лизэнцефалии лежит внутриутробное нарушение миграции нейронов коры. Микроскопически выявляются отсутствие нормальных слоев коры и скопление групп нейронов в субкортикальном белом веществе.
Клинически лизэнцефалия проявляется с рождения или на 1-м году жизни полиморфными судорогами, грубой задержкой психомоторного развития, спастическими параличами и парезами. Уточнить характер патологии можно только при патоморфологическом исследовании.
МЕГ АЛЭНЦЕФАЛИЯ
Мегалэнцефалия — порок развития мозга, при котором глиальные элементы диспропорционально увеличены по сравнению с нейронами. Мегалэнцефалия сопровождается увеличением массы и объема мозга, окружности головы без признаков повышения внутричерепного давления. Встречается с частотой 1 случай на 5000—6000 новорожденных, причем преимущественно у мальчиков.
В клинической картине ведущими являются судороги и задержка психического развития. Мегалэнцефалия может быть следствием не только истинного глиоза, являющегося пороком развития, но и нарушенного обмена веществ при таких формах патологии, как болезнь Тея—Сакса, метахроматическая лейкодистрофия и др.
ПОРОКИ РАЗВИТИЯ ЧЕРЕПНЫХ НЕРВОВ
Аномалии развития черепных нервов проявляются аплазией их ядер, атрофией волокон или их сочетанием. Часто наряду с недоразвитием волокон черепных нервов отмечается нарушение развития костных каналов, через которые они проходят.
Рис. 86. Синдром Мебиуса у детей с множественными пороками развития.
Врожденные пороки развития обонятельного нерва изолированно встречаются редко; отсутствие обонятельных трактов обычно сочетается с лизэнцефалией.
Пороки развития зрительного нерва могут проявляться атрофией его волокон с разрастанием глии на всем протяжении нерва, аплазией или гипоплазией диска, его избыточной миелинизацией и колобомами. Эти пороки бывают односторонними или двусторонними, изолированными или сочетаются с аномалиями развития мозга и глаз. Зрение резко снижено или отсутствует с рождения.
Пороки развития глазодвигательных нервов чаще бывают односторонними и связаны с аплазией ядер или нарушением строения глазницы. Они проявляются сходящимся, расходящимся косоглазием, нарушением зрачковых реакций, птозом. Изолированный врожденный птоз наблюдается при аплазии боковых парных ядер глазодвигательного нерва. При недоразвитии заднего продольного пучка может быть парез взора в сторону или врожденный крупноразмашистый нистагм, сочетающийся с ритмичными движениями головы в сторону.
Аплазия ядер тройничного нерва наблюдается крайне редко. Сохранение эмбриональной связи между двигательными ядрами тройничного нерва и ядрами глазодвигательного нерва обусловливает сочетанные движения глаз и нижней челюсти, известные под названием синкинезии Маркуса—Гунна. Сийкинезия Маркуса—Гунна известна в трех вариантах: 1) непроизвольное поднимание птозированного века при открывании или закрывании рта; 2) поднимание птозированного века при движении челюсти вбок; 3) ассоциация движений век с движениями нижней челюсти.
Врожденные поражения лицевого нерва могут быть связаны с аплазией ядер, атрезией костного канала или шилососцевидного отверстия.
Синдром Мебиуса — врожденная аплазия ядер отводящего и лицевого нервов (рис. 86). Многие случаи этого синдрома спорадические, но имеются описания семей с аутосомно-рецессивным и аутосомно-доминантным наследованием.
Частота синдрома. Мебиуса составляет 1 случай на 20 000 новорожденных.
Лицо ребенка маскообразное, без морщин, с открытым ртом и незакрывающимися глазами. Углы рта опущены. Глазные яблоки не отводятся в стороны. Иногда наблюдается полная наружная офтальмоплегия вследствие аплазии ядер глазодвигательных нервов или полного отсутствия наружных мышц глаза.
Менее постоянно при синдроме Мебиуса отмечается недоразвитие ядер V, VIII, IX, X, XII черепных нервов. Поражение каудальной группы ядер проявляется клиникой бульбарного паралича и быстро приводит к летальному исходу. Пороки развития каудальной группы черепных нервов изолированно встречаются редко, а в сочетании с другими нарушениями обнаруживаются при пороках развития мозгового ствола, синдромах платибазии, базилярной инвагинации, Клиппеля—Фейля и др.
Кроме аплазии ядер, могут наблюдаться другие аномалии развития: микрофтальм, деформации ушей, аномалии прикуса, лишние пальцы и отсутствие пальцев, синдактилия, брахидактилия, косолапость, недоразвитие грудных мышц. Интеллект снижен примерно у 40% больных. Синдром Мебиуса следует дифференцировать от травматических поражений лицевого нерва в родах.
ДИСГЕНЕЗИИ МОЗЖЕЧКА
Пороки развития мозжечка могут выражаться в равномерном недоразвитии всех его отделов, отсутствии червя или его гемисфер, микрогирии коры. Эти пороки часто сочетаются с отсутствием нижних олив и уменьшением размеров основания моста.
У детей с рождения отмечается мозжечковая дисфункция, которая проявляется нарушением координации движений. Более отчетливо признаки недостаточности мозжечка выявляются, когда ребенок начинает тянуться к игрушке и переходит в вертикальное положение. Дети с трудом овладевают навыками сидения, стояния, ходьбы. Походка атактическая, с широко расставленными ногами. В руках выявляются дизметрия, интенционный тремор. Речевые навыки формируются с опозданием из-за дискоординации речевой мускулатуры. Степень выраженности мозжечковых нарушений варьирует в зависимости от тяжести морфологического дефекта. С возрастом наблюдается некоторая тенденция к компенсации мозжечковых расстройств, особенно при целенаправленной тренировке координаторных функций.
Диагноз дисгенезий мозжечка подтверждается данными рентгенологического исследования.
КРАНИОСТЕНОЗ
Краниостеноз — преждевременное закрытие черепных швов, ведущее к ограничению объема черепа, его деформации и повышению внутричерепного давления. Заболевание встречается с частотой 1 случай на 2000 новорожденных, при этом у мальчиков в 2 раза чаще, чем у девочек.
В патогенезе краниостеноза имеют значение обменные нарушения, вызывающие ускоренный остеосинтез в костях черепа, нарушение васкуляризации костей и оболочек в результате воспаления, рентгеновского облучения плода в первой половине беременности.
Преждевременное закрытие коронарного шва ограничивает рост черепа в переднезаднем направлении. Закрытие сагиттального шва влечет за собой уменьшение поперечного размера черепа, уплощение теменных костей. Избыточный рост костей в области поперечных швов приводит к увеличению продольного диаметра черепа. Остроконечная форма черепа формируется в результате преждеврменного закрытия всех швов, сопровождающегося уменьшением продольного и поперечного размеров.
Клиническая картина. Неврологическая симптоматика зависит от степени повышения внутричерепного давления и нарушения венозного оттока из полости черепа. По степени клинического проявления выделяют краниостеноз компенсированный и декомпенсированный. Компенсированная форма краниостеноза характеризуется изменением формы черепа и умеренно выраженными симптомами повышения внутричерепного давления. Дети жалуются на головную боль, однако локальная неврологическая симптоматика отсутствует. На глазном дне определяются умеренно выраженные застойные явления, давление цереброспинальной жидкости 200—300 мм вод. ст. На краниограмме — заращение всех швов, усиление пальцевых вдавлений (рис. 87).
В стадии декомпенсации внутричерепное давление значительно повышено. Почти постоянно наблюдается двусторонний экзофтальм, имеются признаки поражения глазодвигательных нервов, могут возникать генерализованные судорожные припадки. На глазном дне явления застоя и вторичная атрофия зрительных нервов. В большинстве случаев имеет место концентрическое сужение полей зрения на все цвета. Давление цереброспинальной жидкости нередко превышает 500 мм вод. ст. На рентгенограмме черепа черепные швы не дифференцируются, кости свода черепа значительно истончены, выражены пальцевые вдавления, внутренняя поверхность свода черепа приобретает грубый пятнистый рисунок, передняя и средняя черепные ямки деформированы — укорочены и углублены.
Диагноз ставят на основании характерных изменений черепа; его подтверждают результаты рентгенологического исследования и осмотра глазного дна. Краниостеноз дифференцируют от микроцефалии, при которой черепные швы сохранены, объем мозга соответствует объему черепа, нет признаков значительного повышения внутричерепного давления. В стадии декомпенсации следует исключить объемный процесс.
Рис. 87. Краниостеноз.
А. Схема хирургического лечения краниостеноза. Б. Краниограмма больного с краниостенозом (а, б).
Лечение хирургическое, направлено на увеличение объема полости черепа.
АКРОКРАНИОДИСФАЛАНГИЯ (СИНДРОМ АПЕРТА)
Акрокраниодисфалангия — врожденный порок развития черепа, сочетающийся с аномалиями развития кисти. Преждевременное закрытие венечного и стреловидного швов обусловливает башенную форму черепа и приводит к повышению внутричерепного давления. Постепенно формируется вторичная атрофия зрительных нервов. Иногда слепота наблюдается уже при рождении. Вследствие раннего окостенения швов лицевого черепа отмечаются и другие характерные внешние признаки: лунообразное лицо, приплюснутый нос, экзофтальм. Аномалии развития рук проявляются синдактилией, полидактилией, тугоподвижностью в локтевом суставе. Больные отстают в умственном и физическом развитии.
Лечение хирургическое, направленное на увеличение объема черепа, коррекцию синдактилии и полидактилии. Тугоподвижность суставов лечат консервативно. С целью стимуляции психического развития назначают аминалон, церебролизин, пирацетам, ацефен и другие препараты.
Последнее изменение этой страницы: 2016-08-16; Нарушение авторского права страницы
Читайте также: