
Заболевание вильсона коновалова с неврологической симптоматикой
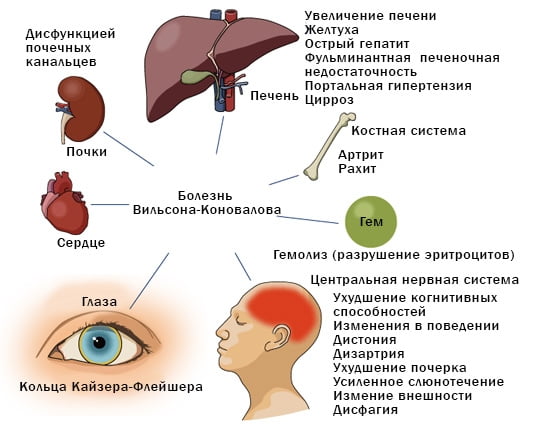
Болезнь Вильсона-Коновалова (синонимы: гепатоцеребральная дегенерация — ГЦД, болезнь Вильсона, гепатолентикулярная дегенерация) — редкое наследственное заболевание ЦНС. Его частота — 1—2 случая на 100 тыс. населения [4]. Впервые описано A. Wilson в 1912 г. Большой вклад в изучение патогенеза и клиники этой патологии внес Н. В. Коновалов [1]. Заболевание названо именем в их честь. Установлено развитие ГЦД вследствие множественных (более 100) мутаций гена ГЦД на 13-й хромосоме, кодирующего синтез медь-транспортной АТФазы. Из-за генетического дефекта выведения медь в больших концентрациях аккумулируется в печени, мозге, почках, роговице, радужной оболочке глаза.
Заболевание наследуется по аутосомно-рецессивному типу. Частота семейных случаев достигает 61% [3]. Различают три генотипических типа ГЦД:
1) славянская (поздняя в 20—35 лет), характеризуется неврологической симптоматикой и незначительным поражением печени;
2) западная (ювенильная в 10—16 лет), отличается первичным поражением печени, и затем появлением неврологической симптоматики;
3) атипичная (проявляется только снижением уровня церулоплазмина без клинических признаков заболевания).
Неврологические проявления ГЦД характеризуются значительным клиническим полиморфизмом. Выделяют пять основных клинических форм заболевания: абдоминальная, ригидно-аритмо-гиперкинетическая, дрожательно-ригидная, дрожательная и экстрапирамидно-корковая формы [1]. Особенность клинической картины ГЦД определяется локализацией поражения в подкорковых ганглиях и коре, а также возрастом начала болезни. Установлено преобладание гиперкинезов при поражении черной субстанции и ригидности — при локализации в бледном шаре. Считается, что при начале болезни до 30 лет чаще встречаются изменения мышечного тонуса и гиперкинезы, а у лиц старше 30 лет — акинетико-ригидный синдром [4]. Чрезвычайный интерес представляет внутрисемейный полиморфизм ГЦД, причины которого в настоящее время остаются неясными.
Мы наблюдали 26 больных с ГЦД (15 мужчин, 11 женщин, средний возраст начала болезни 24,9± 1,5 лет), в 50% случаев установлен наследственный характер болезни. Среди семейных случаев наблюдали брата с экстрапирамидно-корковой и сестру — с дрожательной формой ГЦД. Приводим эти наблюдения.
При осмотре состояние удовлетворительное, обычного питания, отмечается кровоточивость десен, гиперсаливация. АД 130/80 мм рт. ст. Печень у края реберной дуги, безболезненная. В сознании, эйфоричен, снижена критика. Лицо гипомимично. Ослаблена конвергенция, выражены дисфония, дисфагия, дизартрия, глоточный рефлекс высокий, вызываются рефлексы орального автоматизма, отмечаются насильственные эмоции.
Парезов конечностей нет, мышечный тонус повышен по экстра-пирамидному типу. Сухожильно-периостальные рефлексы высокие, с расширенными зонами, D>S, симптом Бабинского справа. Периодически отмечаются миоклонические вздрагивания конечностей и головы. Снижена двигательная инициатива, движения скудные. Походка своеобразная с элементами торсии туловища.
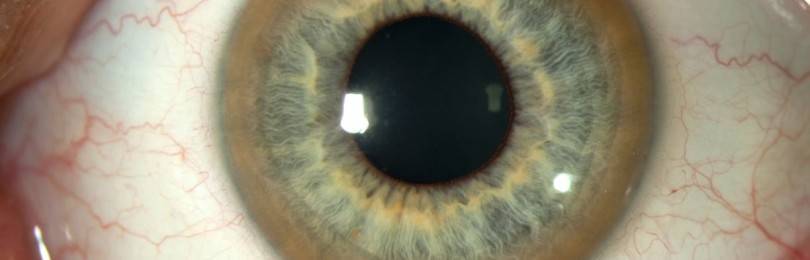
Общеклинические анализы крови и мочи без патологии. Биохимический анализ крови: церулоплазмин 0,1 ммоль/л (норма 1,8-3,5 ммоль/л), общая медь 0,76 ммоль/л (норма 1,0-2,1 ммоль/л). Содержание меди в моче 3,1 ммоль/л (норма 0,1 ммоль/л). Окулист: визус 0,09/0,03, биомикроскопически определяются кольца Кайзера-Флейшера. ЭЭГ: легкая дезорганизация коркового ритма, патологических форм активности не выявлено. МРТ головного мозга: выявлены симметричные участки гиперинтенсивного сигнала в T]W режиме в области чечевицеобразного ядра и ограды (рис. 19). УЗИ брюшной полости: деформированный желчный пузырь, нефроптоз справа.

Рис. 19. МРТ головного мозга в T1w режиме больного Б., 30 лет, с экстрапирамидно-корковой формой ГЦД: симметричные участки гиперинтенсивного сигнала в области чечевицеобразного ядра и ограды (указаны стрелками)
Назначен купренил (до 6 табл/сут). При катамнестическом наблюдении в течение трех лет заболевание приняло стационарное течение. В неврологическом статусе сохраняются умеренные псевдобульбарный и экстрапирамидный синдромы, судорожные приступы и миоклонии не повторялись.
Больная Т., 38 лет, старшая сестра, инвалид II группы, при поступлении жаловалась на дрожание кистей рук, общую слабость, повышенную утомляемость, увеличение живота в объеме, отеки на ногах. Больна в течение трех лет, когда впервые выявлен цирроз печени, который периодически осложняется асцитом и гидротораксом. Неоднократно лечилась в отделении портальной гипертензии. В течение 6 месяцев заметила постепенно нарастающее дрожание кистей. Из ранее перенесенных заболеваний отмечает только простудные. Кроме младшего брата в семье подобным заболеванием никто не болеет. При поступлении общее состояние удовлетворительное, умеренного питания, бледность кожных покровов, иктеричность склер. АД 90/50 мм рт. ст. Живот увеличен в размерах, печень не пальпируется. Умеренная отечность голеней и стоп. Неврологически: в сознании, эмоционально лабильна, снижена оперативная память. Черепные нервы в норме. Сила и мышечный тонус в конечностях не изменены. Сухожильно-периостальные рефлексы живые, зоны расширены, D=S, подошвенные снижены, патологических знаков нет.
В покое и при активных движениях имеет место постоянный, ритмичный, мелкоамплитудный тремор кистей рук. Координа- торные пробы выполняет удовлетворительно, в позе Ромберга устойчива.
Общий анализ крови: НЬ 88 г/л (анизоцитоз, микроцитоз), СОЭ 55 мм/ч. Биохимический анализ крови: билирубин 16,0 ммоль/л, церулоплазмин 0,8 ммоль/л, медь 1,0 ммоль/л. Медь в моче 2,5 ммоль/л. Окулист: острота зрения 0,9/0,9, биомикроскопически определяются кольца Кайзера—Флейшера. Несмотря на проводимое лечение спустя год больная умерла в связи с продолжающейся декомпенсацией цирроза печени и внутрипе- ченочной гипертензией.
В настоящее время установлено, что ГЦД относится к моногенным заболеваниям с аутосомно-рецессивным наследованием. Причины генных мутаций окончательно не установлены, по мнению К. Надировой, связаны с неблагополучным состоянием природы в зонах ядерных полигонов, в регионах цветной и черной металлургии и повышенной радиацией [3], что, возможно, имело место в нашем случае. Развитие заболевания обусловлено нарушением метаболизма меди вследствие первичного биохимического дефекта белков, осуществляющих транспорт меди. Нарушается выведение из печени фракций меди с желчью, снижается скорость включения меди в церулоплазмин. Медь проникает через гематоэнцефалический барьер и вызывает сначала функциональные, а затем структурные изменения в ЦНС. В результате хронической интоксикации организма ионы меди накапливаются в базальных ганглиях головного мозга, печени, роговице, почках [1, 2, 4]. Причины своеобразного патоморфоза и тропизма поражения нервной системы при ГЦД объясняются особенностью ангиоархитектоники подкорковых структур мозга. Капилляры базальных узлов поражаются в результате аноксии больше, чем капилляры в других частях мозга, так как в физиологических условиях в подкорковых узлах отмечается более высокая концентрация меди и интенсивнее ее кругооборот [2].
До развития неврологических симптомов может быть два варианта течения болезни:
1) бессимптомная, когда выявляются только изменения генов;
2) имеет место исключительно абдоминальная симптоматика.
Типичная неврологическая картина чаще развивается после 20 лет с появления экстрапирамидного синдрома. В клинической практике чаще встречается дрожательно-ригидная форма болезни. Экстрапирамидно-корковая форма считается крайне редким проявлением заболевания, и ее появление чаще связывают с выраженной гепатопатией в терминальной стадии заболевания [2].
Диагноз ГЦД подтверждают снижение церулоплазмина и меди в крови (в два раза и больше), увеличение экскреции меди с мочой, повышение содержания меди при биопсии печени. У 70-90% больных в неврологической стадии наблюдаются кольца Кайзера-Флейшера, причем установлена прямая зависимость между возрастом их появления и тяжестью течения ГЦД [1, 2]. Патология печени встречается в 70% случаев ГЦД и чаще имеет латентный характер, но может и предшествовать развитию неврологических симптомов [3].
Основу лечения ГЦД составляет применение Д-пенициламина (купренила), оказывающего медьвыводящее действие, который применяют пожизненно. В комплексное лечение ГЦД целесообразно включать комплексоны (унитиол), антиоксиданты (витамин Е, эссенциале), аминокислоты, нуклеиновые кислоты, витамины группы В. Для успешной терапии ГЦД имеет значение коррекция вторичных нарушений метаболизма с помощью препаратов цинка (цинктерал, сульфат цинка), которые используются как вспомогательное средство с целью снижения дозы купренила [3]. Все больные ГЦД нуждаются в диете и исключении из питания медьсодержащих продуктов (кофе, орехи, бобы, шоколад, грибы, печень). В случаях прогрессирования заболевания проводится трансплантация печени [4].

И.А. Иванова-Смоленская
профеcсор, доктор медицинских наук
ГУ НИИ неврологии РАМН
Болезнь Вильсона–Коновалова (гепато-лентикулярная дегенерация) относится к тяжелейшим наследственным болезням центральной нервной системы и внутренних органов. Если своевременно не начать лечение, направленное на выведение токсичных избытков меди из организма, то через 5–7 лет больной обречен на смерть. Болезнь поражает 25% братьев и сестер в семье при клинически здоровых родителях, которые являются носителями аномального гена (аутосомно-рецессивный тип наследования). Заболевают только те индивидуумы, которые унаследовали два мутантных гена, то есть по одному от матери и от отца – гомозиготные носители мутации; лица, которые от одного из родителей получили мутантный ген, а от другого – нормальный ген, являются гетерозиготными носителями мутации и остаются здоровыми.
Открытый недавно ген болезни отвечает за синтез медь-транспортирующего белка (АТР7В). При гепатолентикулярной дегенерации обмен меди и медьсодержащих белков нарушается, появляется избыток “свободной” меди, которая в больших количествах откладывается в печени, мозге, роговице, а также выделяется с мочой. Не случайно диагностика болезни базируется на обнаружении характерных нарушений медного обмена. Благодаря идентификации гена в настоящее время возможна и ДНК- диагностика этого заболевания.
Поражение печени избытком “свободной” меди проявляется циррозом печени. Поражение мозга приводит к развитию тяжелой неврологической симптоматики: дрожанию конечностей и всего туловища, повышению мышечного тонуса, иногда сопровождающемуся болезненными спазмами, нарушением речи, глотания, снижению интеллекта. Отложение меди в роговице (по краю радужной оболочки) обусловливает формирование кольца Кайзера–Флейшера – буро-зеленоватого пигмента. По этому признаку диагноз болезни можно поставить безошибочно.
Гепато-лентикулярная дегенерация известна с глубокой древности. Дошедшее до нас изображение египетского фараона Тутанхамона, по мнению крупнейшего специалиста J. Walshe, не исключает вероятности, что он страдал этим заболеванием. Институт неврологии РАМН в течение многих лет занимается проблемой гепато-лентикуляной дегенерации. Знаменитый отечественный невролог академик АМН СССР Н.В. Коновалов, один из основателей Института, посвятил этому заболеванию две монографии, последняя из которых в 1964 году была удостоена Ленинской премии. В последующие годы данное заболевание продолжало успешно изучаться сотрудниками нейрогенетического отделения Института неврологии, под наблюдением которых за 40 лет находилось свыше500 семей, отягощенных этим недугом. Весь многолетний опыт Института свидетельствует о том, что ключевой проблемой является ранняя диагностика гепатолентикулярной дегенерации. Чем раньше начать лечение (в идеале – еще на досимптоматической стадии либо на доневрологическом этапе, то есть до появления признаков поражения мозга), тем лучше эффект. Вот почему, если в семье есть хоть один ребенок, страдающий этим заболеванием, необходимо тщательное обследование всех его братьев и сестер, в том числе с использованием самых современных биохимических и молекулярно-генетических методов.
Заподозрить раннюю стадию болезни можно на основании следующих признаков: перенесенной желтухи; повторных кровотечений из носа, кровоточивости десен либо множественных кровоподтеков; сосудистых “звездочек” на коже груди и спины; своеобразных “полосок” (белых, меняющих периодически окраску на красновато-синюшную) на бедрах и в подмышечных областях; гормональных нарушений в виде аменореи или дисменореи у девушек, гинекомастии (нагрубание грудных сосков) у юношей, а также акромегалии(увеличение носа, подбородка, утолщение губ); снижения интеллекта и изменений психики в виде чередования дурашливости и пониженного настроения, трудностей усвоения нового материала, проблем с успеваемостью в школе.
Гепато-лентикулярная дегенерация может начать проявляться в детском, подростковом, юношеском, зрелом возрасте и очень редко – в 50–60 лет. Чем раньше начинается заболевание, тем тяжелее оно протекает (при отсутствии лечения). Однако болезнь Вильсона–Коновалова – редкий пример наследственного нарушения, для которого разработаны высокоэффективные методы лечения: даже при появлении тяжелой неврологической симптоматики систематическое лечение обычно дает “драматический” эффект, вплоть до исчезновения всех симптомов или резкого их уменьшения. Пациенты вновь могут полностью обслуживать себя, вести домашнюю работу, учиться, работать по профессии, создать семью и родить здорового ребенка (под нашим наблюдением находятся 30 женщин, страдающих гепатолентикулярной дегенерацией и благополучно родивших здоровых детей). Пациентам с гепатолентикулярной дегенерацией необходимо регулярно наблюдаться у постоянного лечащего врача.
В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин.
Эти препараты назначаются по специальной схеме с постепенным увеличением дозы. К сожалению, в силу необходимости проведения пожизненного лечения и особых требований к химической чистоте препаратов отечественный аналог пеницилламина не может быть рекомендован при гепатолентикулярной дегенерации из-за высокой токсичности.
При длительном многолетнем приеме D-пеницилламина у некоторых больных гепатолентикулярной дегенерацией возникают побочные явления в виде дерматитов, анемии и иных осложнений. Поэтому был предложен альтернативный метод лечения солями цинка (оксид, сульфат и др.). Нами было предложено комбинированное лечение D-пеницилламином и препаратами цинка, что дает возможность снизить дозу и избежать побочных явлений. У больных в пресимптоматической стадии достаточно лечения только препаратами цинка.
В настоящее время за рубежом в тяжелых случаях болезни, не поддающихся консервативному лечению, широко применяется пересадка печени. При удачном исходе операции больной полностью выздоравливает и не нуждается в дальнейшем приеме препаратов. В России делаются первые шаги в этом направлении, и одним из таких шагов является разработанный нами совместно с Институтом трансплантологии и искусственных органов метод био-гемоперфузии с изолированными живыми клетками печени и селезенки – так называемый аппарат “вспомогательная печень”.
Помимо этих методов, большое значение имеет гепатопротекторная терапия, направленная на максимальное улучшение функций печени.
Таким образом, при правильной терапии гепатолентикулярной дегенерации – тяжелейшего наследственного заболевания мозга и внутренних органов – в 80% случаев возможно клиническое выздоровление либо выраженное улучшение состояния больных при условии своевременной максимально ранней диагностики.
Гепатолентикулярная дегенерация (другое название синдром Вильсона-Коновалова) – это редкое и сложное заболевание, которое развивается вследствие тяжелых метаболических расстройств в организме. Имеет наследственную природу. Встречается у одного человека на 100 тысяч.
Впервые недуг описал Сэмюель Вильсон в далеком 1912 году. В России большой вклад в изучение патологического процесса внес невролог Коновалов. Он создал классификацию заболевания. Ее используют и сейчас при формулировке клинического диагноза.
Чаще всего диагностируют в возрасте 5-45 лет. Хотя встречаются клинические случаи, когда патология проявлялась у маленьких детей либо пожилых людей. Рассмотрим патогенез, симптомы, формы и способы лекарственной терапии.
Этиология синдрома Вильсона-Коновалова

Заболевание носит генетическую природу. Синдром Вильсона-Коновалова и синдром Вильсона-Микити – это разные недуги. В последнем случае речь идет о патологии легких, которую диагностируют только у новорожденных детей. Проявляется развитием поздней кислородозависимости.
Причина формирования патологии кроется в генетике. Ген, который несет ответственность за возникновение недуга, располагается в 13-й хромосоме. Он принимает участие в перемещении меди. Заболевание Вильсона наследуется от родителей как рецессивный аутосомный признак, развивается вследствие даже незначительной мутации гена.
Особенность недуга в том, что заболеет ребенок только в том случае, если мама и папа являются носителями гена. Люди, имеющие в анамнезе один пораженный ген, не страдают от симптоматики патологии, но у них могут быть незначительные нарушения метаболизма меди.
В организме взрослого человека средняя концентрация меди около 100 мг. При этом потребность в сутки для нормального функционирования всех органов и систем 1-2 мг. На фоне чрезмерного поступления вещества в организм излишки всасываются печенью, вместе с желчью выводятся естественным путем.
При болезни Вильсона нарушаются сразу два процесса – это биологическая выработка белков, которые связывают желчь, и выведение естественным способом. Вследствие этого содержание вещества возрастает, медь откладывается в органах:
- Роговицы глаз.
- Почки.
- Печень.
- Головной мозг.
При критическом возрастании концентрации происходит токсическое поражение внутренних органов человека. Развивается цирроз печени, чаще всего крупноузловой формы. В головном мозге проявляется нарушение функциональности мозжечка, а в глаза формируется кольцо Кайзера-Флейшера/
Единственная причина формирования недуга – мутация определенного гена, который несет ответственность за метаболизм меди. В данном носителе ученые обнаружили более 100 возможных отклонений, поэтому анализ вероятных нарушений ДНК – неэффективный способ.
Предупредить недуг невозможно, профилактики не существует. Если ген имеется у обоих родителей, то у ребенка уже к 2-3 годам будут проблемы с нарушением работы печени.
Симптоматика

Гепатолентикулярная дегенерация имеет код по МКБ Е83.0. При клиническом описании патологического процесса медицинские специалисты обязательно указывают вид недуга, выраженность нарушений со стороны центральной нервной системы, печени.
Клиника развивается в раннем возрасте. Симптомы похожие на различные болезни железы. Чаще всего маленькие пациенты страдают от выраженного пожелтения кожных покровов, астении, анорексии. У девочек часто повышается температура тела. Сбить лекарственными средствами не получается.
Печень больного насыщается медью. После вещество начинает скапливаться в других органах, что приводит к сбою в работе ССС, ЖКТ, ЦНС. В последнем случае скопление меди в нервной системе негативно влияет на моторику, мимику, координацию движений.
Интеллект у ребенка полностью сохраняется не на фоне всех форм. Еще болезнь влияет на поведение – проявляются агрессивность и раздражительность.
Когда в глазах увеличивается концентрация меди до критического уровня, то происходит формирование кольца коричневого цвета. Его можно диагностировать посредством щелевой лампы, но только у детей старше 5-летнего возраста.
Гепатолентикулярная дегенерация сопровождается клиническим полиморфизмом, в процесс вовлекаются органы выделительной системы. У патологии присутствуют рецессивные признаки, которым предшествуют расстройства ЖКТ.
Виды заболевания и их симптомы
В зависимости от клинических проявлений в медицинской практике выделяют три формы патологии Вильсона-Коновалова – болезнь, которая нарушает работу печени, заболевание, поражающее ЦНС, смешанный вид. Согласно типам у больного преобладает та либо иная клиника.
Печеночный тип иногда называют брюшным. Он развивается у людей до 40-летнего возраста, сопровождается расстройством работы печени. Симптоматика схожа с клиническими признаками циррозного поражения.
В 80% клинических картин печеночный тип у пациентов протекает с проявлением таких симптомов:
- Слабость, постоянная усталость.
- Повышенное газообразование.
- Боль в области печени тупого характера.
- Увеличение объема жидкости в брюшной полости.
- Из носа периодически идет кровь.
- Пожелтение кожного покрова.
- Лихорадочное состояние.
- Увеличивается в размере селезенка.
- Утолщаются пальцы верхних/нижних конечностей.
В остальных 20% клинических картин к этим симптомам могут добавляться другие признаки заболевания.
Данный вид патологического процесса отличается другими проявлениями. Они начинают развиваться в раннем возрасте. У детей наблюдается мышечная ригидность, расстройство речи, постепенно снижаются интеллектуальные способности. На фоне неврологического типа бывают периоды обострения и затишья.
При обострении патологии в 10-25 лет у больных проявляется тремор конечностей, нарушение темпа речи из-за затруднения издавать расчлененные звуки. Пациенты медленно пишут, читают, разговаривают, часто бесцельно двигают руками.
Любой вид синдрома Вильсона-Коновалова может дополниться другой симптоматикой. Она выявляется в 20% случаев. Симптомы – увеличение грудных желез у мужчин, нарушение слухового восприятия, чрезмерная хрупкость костей, что приводит к постоянным травмам, посинение ногтевых пластин, кожных покровов. Также нарушается работа почек, проявляется гемолитическая форма анемии.
В соответствии с общепринятой классификацией заболевание бывает острого и хронического течения. Врачи также выделяют еще скрытую стадию, длительность которой не более 7-летнего периода. На фоне латентной стадии симптоматика имеется, однако она не оказывает существенного влияния на качество жизни больного.
В некоторых случаях патология никак не проявляет себя до 5-летнего возраста ребенка. Пик заболевания приходится на возраст 8-15 лет, однако уже с рождения имеются нарушения работы печени.
В таблице представлены особенности заболевания в зависимости от типа течения:
| Тип течения | Описание |
| Острый | Заболевание ярко проявляется уже в раннем возрасте, симптомы острые, выражены сильно, возникают комплексно и сразу. Организм пациента быстро угасает, медикаментозная терапия не приносит облегчения. Риск летального исхода при такой клинике свыше 90%. Несмотря на развитость медицинской науки, способов помочь таким пациентам еще не придумали. |
| Хронический | Отличается этот вид медленным прогрессированием. Вначале может быть скрытая стадия. Сначала страдает печень, после ЦНС. В подростковом возрасте имеются проблемы с координацией движения. Выраженность симптоматики средняя, прогрессирует клиника медленно. |
Методы диагностики
Диагностикой патологии занимаются несколько врачей – гепатолог, нефролог и гастроэнтеролог. Так как болезнь связана с наследственным фактором, то в диагностический процесс привлекают генетиков. Также требуются профессиональные консультации дерматолога, офтальмолога и ряда других специалистов, чтобы выявить осложнения.
Для начала собирают анамнез больного, осуществляют физикальный осмотр. На основании результатов дают клинические рекомендации, назначают соответствующие методы диагностики.
Диагностика включает в себя исследование урины и крови (определяют уровень меди). У офтальмолога с помощью щелевой лампы определяют отсутствие/наличие кольца Кайзера-Флейшера. Дополнительно проводится УЗИ, МРТ головного мозга, забор биоптата из печени.
Способы лечения

Терапия ориентирована на уменьшение поступления вещества в организм пациента. Для этого рекомендуется исключить из рациона продукты, которые содержат медь. Из меню убирают баранину, свинину, кальмары, креветки, крабов, грибы, сухофрукты, шоколад. Можно кушать молочные продукты, фрукты, овощи, курицу, куриные яйца, хлебобулочные изделия.
Чтобы снизить содержание меди в организме, когда оно неуклонно растет, медицинские специалисты рекомендуют прием медикаментов противовоспалительного, иммуносупрессивного свойства. В схему терапии входят лекарства желчегонного свойства, антиоксиданты, препараты с цинком.
Дозировка пациенту устанавливается индивидуально. Обычно назначают самую маленькую и отслеживают динамику улучшений. Дополнительно рекомендуется прием витаминно-минеральных комплексов. Перед покупкой оных нужно внимательно изучать инструкцию, в составе не должно быть меди.
Если медикаментозное лечение не дает нужного эффекта, самочувствие больного ухудшается, то единственный способ – это трансплантация печени. После хирургического вмешательства продолжают прием поддерживающих медикаментов.
Лечение лучше начинать сразу после выявления заболевания, даже когда выраженные симптомы отсутствуют. Это позволит предупредить клинику, способствует замедлению прогрессирования недуга.
К наиболее распространенному осложнению относят патологии печени. Они проявляются симптомами:
- Пожелтение кожи.
- Деформация пальцев верхних и нижних конечностей.
- Расширение вен на передней брюшной стенке.
- Отечность ног.
Часто у больных выявляется кровотечение в ЖКТ. Развивается печеночная недостаточность, к симптоматике которой относят нарушение сна, расстройства поведения, в тяжелых случаях наблюдается печеночная кома.
К осложнениям относят нарушения неврологического характера. Это мышечная дистония, дизартрия, расстройства поведения, личности, эпилептические припадки. У женщин нарушается фертильность.
Прогноз и профилактические мероприятия
Благоприятный прогноз возможен только в одном случае – если заболевание обнаружили в раннем возрасте, сразу же начали терапию высокоэффективными медикаментами. Важно начать терапевтический курс до поражения ЦНС и печени.
Прием медикаментов улучшает функциональность печени, нивелирует негативную симптоматику со стороны ЦНС. Уже спустя 6 месяцев от начала терапии значительно улучшается самочувствие больного. Более выраженные улучшения видны через 2-3 года.
Если лечение отсутствует, начато поздно либо эффективность терапии недостаточна, то летальный исход наступает в 35-40-летнем возрасте. Как правило, причиной выступает печеночная недостаточность. При серьезных поражениях железы требуется трансплантация органа. Чем раньше ее сделать, тем лучше приживется орган. Выживаемость среди больных 20-ти лет составляет 80%.
Специфических профилактических мер нет, потому что болезнь связана с наследственностью человека. Тем, кто находится в группе риска, рекомендуется периодически посещать врача, проходить обследования, заниматься спортом, полностью отказаться от употребления алкогольной продукции.

Проявления этой болезни еще в начале XX века описал английский врач Вильсон (Уилсон), а спустя 50 лет русский невропатолог Коновалов более детально изучил и описал эту патологию, выделил различные формы болезни и предложил ее второе название — гепатоцеребральная дистрофия.
Медь в организме человека присутствует в очень маленьком количестве, однако выполняет важнейшие функции. В норме ее содержание не должно превышать 24 мкмоль/л (у беременных женщин концентрация этого элемента может возрастать почти в 2 раза – это физиологические изменения).
Когда количество этого микроэлемента в организме повышается, он начинает накапливаться во внутренних органах, вызывая тяжелейшие повреждения.
Формы и симптомы болезни Вильсона – Коновалова
Симптомы заболевания проявляются в молодом или зрелом возрасте. Существует несколько форм заболевания. Каждая из форм характеризуется преимущественным поражением той или иной системы организма человека.
Встречается чаще всего: 50-80% случаев. Развивается либо как хронический гепатит, либо как цирроз печени. Такую форму заболевания очень сложно лечить. Характерные симптомы болезни Вильсона-Коновалова:
- Желтуха
Существует три вида желтухи: печеночная, механическая, гемолитическая.
- Механическая желтуха появляется при закупорке выводных желчных протоков. Цвет кожи приобретает зелено-желтый оттенок. Также характерным симптомом является кожный зуд. Желтуха очень сильно выражена.
- Гемолитическая желтуха появляется при повышенном разрушении эритроцитов (красных телец крови). Цвет кожи при этом становится бледно-лимонным. Обычно данный вид желтухи не интенсивен. Кожный зуд отсутствует.
- Печеночная желтуха. Данная форма желтухи проявляется при болезни Вильсона-Коновалова. Печеночная желтуха характеризуется поражением клеток печени, вырабатывающих желчь, что ведет к попаданию билирубина в кровь. Цвет кожи при данной форме желтухи становится желто-оранжевым. Интенсивность печеночной желтухи – умеренная. Кожный зуд появляется редко. Характерными признаками печеночной желтухи служат моча темного цвета (коньячного) и кал белого (бесцветный).
При печеночной желтухе в крови повышается связанный билирубин. Этот билирубин легко выделяется с мочой. Именно поэтому моча приобретает коньячный цвет. Как было описано выше, разрушаются клетки печени, что ведет к снижению выработки желчи. Именно метаболиты желчных кислот окрашивают кал в нормальный цвет. А раз желчь не вырабатывается, поэтому и кал бесцветный.
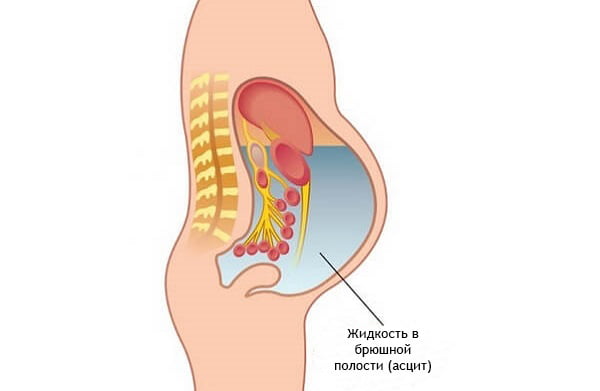
- Асцит
Асцит – скопление в брюшной полости жидкости. Этот симптом появляется в данном случае при значительном поражении печени. Жидкость, похожая по составу на плазму крови, скапливаясь в брюшной полости, давит на все органы, которые там расположены.
У пациента с выраженным асцитом увеличен в размере живот. Причем при смене положения тела увеличенный живот тоже меняет свое положение. Жидкость двигается под действием силы притяжения земли. Жидкость всегда двигается в место, наиболее приближенное к поверхности земли.
- Отёки
Отёки появляются чаще всего на ногах. При поражении почек отеки возникают под глазами.
- Кровотечения
Кровотечения происходят из-за нарушения свертываемости крови. Печень вырабатывает различные факторы свертывания. При болезни Вильсона клетки печени разрушаются, поэтому печень не способна вырабатывать факторы свертывания крови.
- Аменорея
Аменорея – это отсутствие у женщин менструальных циклов. Печень помимо всего еще и инактивирует различные гормоны. В норме печень инактивирует избыток эстрогенов. В данном случае печень не выполняет этой функции, тем самым вызывая аменорею.
Характерными симптомами заболевания являются:
- Тремор рук, головы. Тремор – непроизвольные быстрые движения рук или головы. Для тремора характерна ритмичность, небольшой объем (размах) движений. Тремор может быть постоянным или периодическим (например, отсутствие тремора во время сна).
- Гримасничанье – преувеличенные быстро меняющиеся движения лицевых мышц, возникающие непроизвольно.
- Нарушения почерка. Почерк становится неровным, неразборчивым. При написании какого-либо текста наблюдается неодинаковые по размеру и расположению буквы в слове (одни буквы меньше – другие больше, одни выше – другие ниже).
- Дизартрия – расстройство речи, при котором происходит нарушение произношения звуков, слов. Речь становится непонятной, “говорит, как с кашей во рту”. Данное расстройство возникает вследствие нарушения иннервации органов, участвующих в создании речи (мягкого нёба, языка, губ).
- Более поздними симптомами являются: контрактура при сгибании и мышечная ригидность. Контрактура – ограничение пассивных и движений в различных суставах. В данном случае ограничивается разгибательные движения в суставах рук и ног. Мышечная ригидность – повышенный тонус мышц. Из-за увеличенного тонуса различных групп мышц происходит увеличение сопротивления при осуществлении движений (труднее совершить то или иное движение).
- Нарушение психики встречается в 20% случаев. Среди нарушений можно отметить психоз, депрессию. Психоз – психическое расстройство, проявляющееся крайне неадекватным поведением, с присутствием таких явлений, как галлюцинации и бред. Депрессия – расстройство, при котором происходит значительное и стабильное снижение настроения, утрата способности радоваться, потеря интереса к жизни, двигательная заторможенность.
Симптомом, характерным для обеих форм болезни Вильсона, служит Кольцо Кайзера – Флейшера. Он встречается в 50-62% случаев. Кольцо Кайзера – Флейшера – это кольцо коричневатого цвета, которое расположено на наружном крае роговицы глаза. Оно появляется из-за накопления в роговице меди.
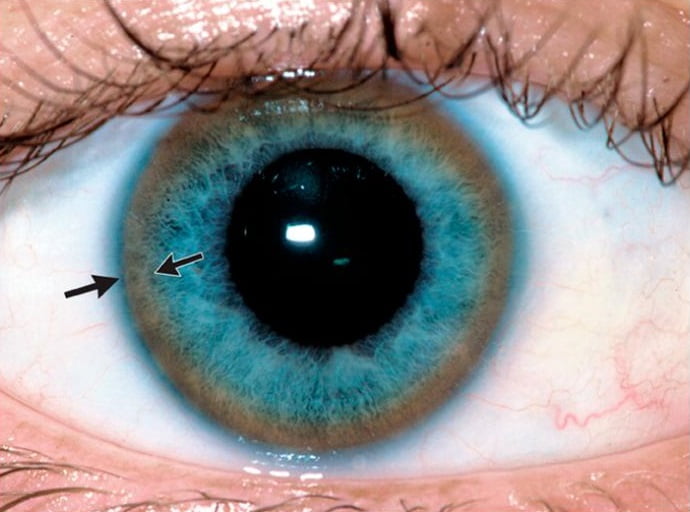
Интересно, что хорошо выраженное кольцо можно увидеть невооруженным глазом. Если интенсивность кольца невелика, чтобы его определить, необходимо специальное оборудование.
- Изменения со стороны почек встречается в 10% случаев. Они случаются из-за накопления меди в клетках почек. Чрезмерное накопление меди ведет к разрушению клеток почки, в результате чего появляются следующие симптомы: Гематурия – присутствие в моче элементов крови (эритроцитов). В норме в моче нет крови. Гликозурия – присутствие в моче глюкозы. В норме в моче не должно быть глюкозы.
- Внутрисосудистый гемолиз – разрушение эритроцитов в полости сосудов. Встречается в 10-15:% случаев. Гемолиз в конечном итоге приводит к анемии (снижение количества эритроцитов и гемоглобина в крови).
- Поражение костной системы – встречается в 20% случаев. Одним из главных проявлений является остеопороз. Остеопороз – заболевание, при котором снижается плотность, и нарушается структура костей. Вследствие остеопороза повышается ломкость костей, что в свою очередь ведет к частым переломам.

Диагностика заболевания Вильсона-Коновалова
- Начальным этапом диагностики болезни Вильсона-Коновалова является физикальное обследование. Уже во время него вероятным признаком заболевания оказывается типичный для данной болезни симптом – кольцо Кайзера-Флейшера. Оно представляет собой жёлто-коричневую обводку по периферии роговицы глаза.
- Следующий этап диагностики болезни Вильсона-Коновалова – лабораторные исследования крови и мочи пациента. Предварительный диагноз подтверждает обнаружение повышенного уровня печёночных ферментов и суточного выделения меди в урине.
- К инструментальным методам диагностики болезни Вильсона-Коновалова относят УЗИ, МРТ и КТ. Благодаря им визуализируется увеличение печени и селезёнки (гепато- и спленомегалия), а также разрушение подкорковых нейронных узлов в головном мозге.
- Генетическая часть диагностики болезни Вильсона-Коновалова заключается тестировании крови пациента и ближайших его родственников на предмет обнаружения патологического гена.
- При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца.
Лечение болезни Вильсона
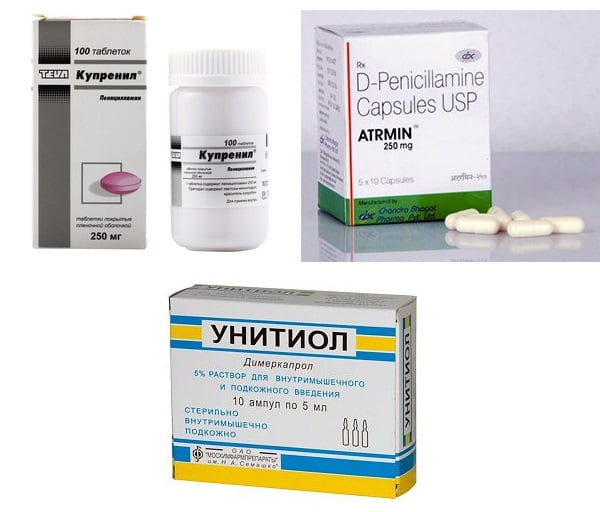
Основой патогенетического лечения болезни Вильсона является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола.
Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза).
Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов).
В случае доминирования гиперкинезов рекомендуется назначение небольших курсов нейролептиков, при ригидности (повышенном тонусе мышц) прописывают леводопы и карбидопы, тригексифенидил (препараты, назначаемые при болезни Паркинсона).
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме.
В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень).

Немедикаментозное лечение болезни Вильсона состоит в назначении диеты (рекомендация – стол №5 с шестиразовым дробным питанием, в котором преоболадает прием белков, ограничен прием липидов и соли) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.)
Прогноз и профилактика болезни Вильсона
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
Читайте также: