
Железо в базальных ганглиях
а) Терминология:
1. Сокращения:
• Пантотенаткиназа-ассоциированная нейродегенерация (ПКАН)
• Инфантильная нейроаксональная дистрофия (ИНАД)
• Нейродегенерация с отложением железа в головном мозге (НОЖГМ)
2. Определение:
• Группа нейродегенеративных нарушений, характеризующаяся дистонией, паркинсонизмом и мышечной спастичностью:
о Вызывается мутациями в гене L-ферритина FTL1
о Все мутации приводят к аномальному накоплению Fe в базальных ганглиях
о Включает ПКАН, ИНАД, ацерулоплазминемию и т.д.:
- Тельца Леви, набухание аксонов, гиперфосфорилированные тау-белки при некоторых подтипах
б) Визуализация:
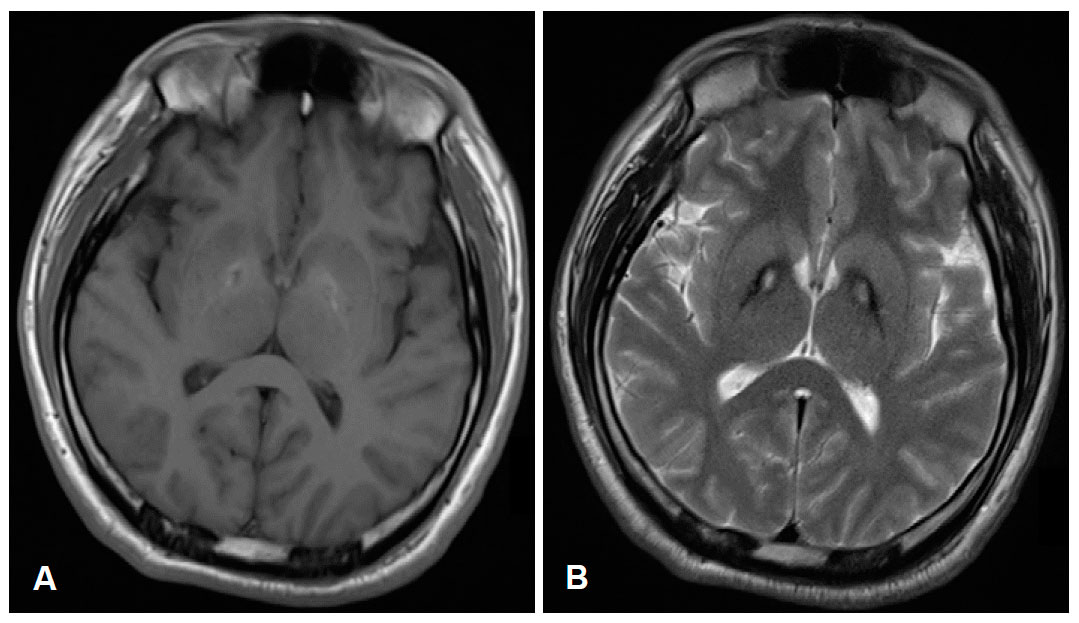
1. Общие характеристики нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Лучший диагностический критерий:
о Снижение интенсивности сигнала на Т2-ВИ от бледных шаров (БШ)
• Локализация:
о ПКАН и ИНАД:
- БШ, черная субстанция (ЧС) ± зубчатые ядра (ЗЯ)
о Нейроферритинопатия и ацерулоплазминемия:
- БШ, ЧС, ЗЯ, кора, полосатое тело и таламус
2. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТс Т2-ВИ* (градиентноеэхо) или изображения, взвешенные по восприимчивости магнитного поля (SWI)
3. КТ признаки нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Бесконтрастная КТ:
о Атрофия больших полушарий и мозжечка при PANK2-негативной нейродегенерации с отложением железа в головном мозге (НОЖГМ)
в) Дифференциальная диагностика нейродегенерации с отложением железа в головном мозге (НОЖГМ):
1. Нормальные процессы отложения железа:
• Визуализируется на ЗТ
• Наблюдается при нормальном процессе старения
3. Рассеянный склероз (PC):
• Накопление железа в базальных ганглиях связано с PC
• Должны быть другие классические демиелинизирующие поражения
4. Поверхностный сидероз:
• Перегрузка железом вследствие переливания крови или рецидивирующих кровоизлияний в ЦНС
5. Гемохроматоз:
• Поражение печени и селезенки обычно происходит до поражения ЦНС
г) Патология:
1. Общие характеристики нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Генетика:
о Аутосомно-рецессивные мутации генов PANK2, PLA2G6 и СР
о Аутосомно-доминантная мутация гена FTL
2. Макроскопические и хирургические особенности:
• Накопление железа, пигментация цвета ржавчины
д) Клиническая картина:
1. Проявления нейродегенерации с отложением железа в головном мозге (НОЖГМ):
• Наиболее частые признаки/симптомы:
о Атаксия, дизартрия, дистония
о Дегенерация сетчатки и атрофия зрительных нервов
• Другие признаки/симптомы:
о Ацерулоплазминемия: дебют во взрослом возрасте с триадой, включающей диабет, дегенерацию сетчатки и двигательные нарушения
о Нейроферритинопатия: дебют во взрослом возрасте с хореей или дистонией
2. Демография:
• Возраст:
о ПКАН: классический вариант- возраст
Редактор: Искандер Милевски. Дата публикации: 25.4.2019

Болезнь Галлервордена-Шпатца ( ригидность прогрессирующая ) – редкое наследственное нейродегенеративное заболевание, при котором происходит поражение базальных ганглиев, и накопление в них железа.
Накопительные процессы происходят в черной субстанции и в бледном шаре. Также в коре головного мозга наблюдаются изменения в нервных клетках, появление пигментированных нейроглий, и глиальных клеток, сходных с глиями при болезни Альцгеймера.
Впервые болезнь была описана немецкими исследователями Юлиусом Галлерворденом и Гуго Шпатцем, именами которых и была названа.
Однако в современной международной медицинской практике болезнь называют нейродегенерацией с отложением железа в головном мозге (NBIA). Отказ от первоначального названия связан с тем, что Юлиус Галлерворден вел исследования эвтаназии в фашистской Германии.
Заболевание считается редким, на 1 миллион человек приходится до 3 человека, имеющих болезнь. Данная нейродегенерация может приводить к паркинсонизму, дистонии, деменции, смерти.
Особенности генетического сбоя
Болезнь Галлервордена-Шпатца это наследственная генетическая патология. Заболевание может быть как семейным, так и спорадическим (случайным), однако всегда передается по наследству. Болезнь относится к аутносомно-рецессивному типу наследования.
Для передачи болезни оба родителя должны быть гетерозиготными носителями заболевания и иметь одну мутировавшую аллель.
Болезнь вызывается мутацией в гене пантотенат-киназы (PANK2) который находится в 20 хромосоме в локусе 20р12.3–р13. Ген пантотенат-киназы отвечает за кодирование протеина пантотенат-киназы 2, который в свою очередь сдерживает процесс накопления аминокислоты цистеина и пантетеина. 
В результате происходит образование химических соединений с ионами железа, которые имеют негативное воздействие на белки и дают старт процессам перекисного окисления, что приводит к программируемой клеточной гибели нейронов. Вместо отмерших нейронов нарастает глиальная ткань (вспомогательные клетки нервной ткани).
Патологические процессы происходят в бледном шаре, красных ядрах и черном веществе (субстанции nigra). В них скапливается субстанция зеленовато-коричневого цвета, которая содержит железо. Помимо этого в белом веществе головного мозга, коре головного мозга, периферических нервных стволах и спинном мозге наблюдаются сфероидные нейроаксональные образования (расширения аксонов с разрастанием тубулярных и мембранных структур).
Формы патологии
У синдрома Галлервордена — Шпатца выделяют три формы. Деление основано на возрасте, в котором начинается развитие заболевания:
- Детская форма. Начало развития болезни происходит в возрасте до 10 лет.
- Ювенильная (подростковая) форма. Болезнь развивается в период от 10 до 18 лет.
- Взрослая (атипичная) форма. Болезнь манифестирует после 18 лет.
Особенности клинической картины
Признаки данной патологии могут варьироваться в каждом конкретном случае, и проявление симптомов зависит от формы болезни у индивида.
Детская форма Галлервордена Шпатца проявляется в возрасте между 5 и 10 годами. Считается классической формой болезни.
В подавляющем количестве случаев (90%) заболевание начинается с торсионной дистонии, которая поражает мышцы ног. У больного происходит сокращение мышц, затрудняется ходьба, изменяется походка. Затем поражаются мышцы лица, глотки и туловища.
Возможно присутствие блефароспазма, спазма кистей, спастической кривошеи, лицевого гемиспазма. Треть пациентов имеют мышечную ригидность, гипокинезию, тремор (признаки синдрома паркинсонизма).
К типичным признакам детской формы болезни также относят:
- эпилептический синдром;
- агрессивность;
- умственную отсталость (возникает из-за ухудшения памяти и внимания);
- асоциальность;
- зрительные нарушения (атрофия зрительных нервов, дегенерация сетчатки);
- психические расстройства.
Подростковая форма развивается и протекает более медленно. В начале болезни проявляются:
- фокальная торсионная дистония (чаще всего затрагивает мышцы конечностей и челюстно-ротовые мышцы);
- нейропсихологические расстройства;
- поведенческие расстройства;
- интеллектуальные расстройства.
Атипичная форма болезни – наиболее редкая (15% случаев заболевания), и протекает отлично от первых двух форм. Для этой формы наиболее характерны такие симптомы:
- синдром паркинсонизма с невозможностью сохранять равновесие в положении стоя;
- непроизвольные движения в разных частях тела, дистония, хорея и атетоз (замедленные и порывистые движения), гемибаллизм (размашистые движения), миоклония (не длительные мышечные спазмы);
- деменция;
- эпилептический синдром;
- депрессивность, агрессия;
- патологические рефлексы, которые возникают вследствие разрушительных процессов в нейронах головного мозга.
Диагностические критерии и методики
Основные критерии для диагностики:
- начало болезни до 30 лет;
- типичная картина в результатах МРТ;
- постоянное развитие симптомов;
- экстрапирамидальные синдромы;
- пирамидные знаки;
- эпилептические приступы;
- когнитивные нарушения;
- пигментная дегенерация сетчатки;
- наличие болезни в семейном анамнезе.

Болезнь необходимо дифференцировать с другими заболеваниями, которые имеют сходную клиническую картину: хореей Гентингтона, болезнью Вильсона-Коновалова, хореоакантоцитозом, болезнью Мачадо-Джозефа.
Для диагностирования пациент направляют на такие обследования:
Чем могут помочь современные врачи?
В современной медицине не существует методов лечения, которые могут предотвратить или остановить болезнь Галлервордена-Шпатца.
Терапия направлена на облегчение и снятие интенсивности симптоматики:
- При синдроме паркинсонизма назначают агонисты дофамина (Проноран, Прамипексол, Мирапекс, Пирибедил), амантадины
![]()
(Симметрел, Мидантан). Однако часто наблюдается резистентность синдрома к лечению. - Для купирования гиперкинезов используют вальпроаты (Конвулекс, Депакин, Энкорат), бензодиазепины (Клоназепам, Диазепам).
- Для снятия спастичности мышц применяются миорелаксанты (Мидокалм, Баклофен).
- Эпилептические приступы снимаются вальпроатами, Томапаксом.
- При когнитивных нарушениях применяют Глиатилин, Нейромидин.
- Для лечения психических нарушений рекомендуют прием нейролептиков (Клоназепама, Кветиапина, Рисполента), антидепрессантов (Дапоксетина, Циталопрама, Венлафаксина).

Появляются и новейшие методы лечения болезни. К ним относятся терапия путем введения пантотеновой кислоты, магнитная стимуляция головного мозга (бледного шара).
Печально, но не безнадежно
При болезни Галлервордена-Шпатца симптомы постоянно прогрессируют. Наиболее тяжело протекает детская форма заболевания. Полная инвалидизация индивида наступает через 10-15 лет с момента первого проявления клинических проявлений.
Наиболее благоприятное развитие болезни прогнозируется при взрослой форме болезни. Особенно в случае слабой выраженности деменции.
Терапия позволяет сохранить относительное качество жизни пациента и его способность к самообслуживанию. Продолжительность жизни при атипичной форме болезни Галлервордена-Шпатца может составлять более 20 лет.
Болезнь Галлервордена — Шпатца – нейродегенеративное заболевание, которое сопровождается накапливанием железа в базальных ганглиях.
| МКБ-10 | G23.0 |
|---|---|
| МКБ-9 | 333.0 |
| DiseasesDB | 29462 |
| MeSH | D006211 |
| OMIM | 234200 |
| MedlinePlus | 001225 |
| eMedicine | neuro/151 |
Содержание
- Общая информация
- Причины и патогенез
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Профилактика
Общая информация
Базальные ганглии – нейронные подкорковые узлы, находящиеся в центральном белом веществе полушарий. Они состоят из полосатого тела, стриатума и чечевицеобразного ядра. В структуру последнего входят бледный шар и скорлупа. Кроме того, к базальным ганглиям относится часть черной субстанции – звена экстрапирамидной системы.
Предназначение базальных ганглиев:
- регуляция двигательных и вегетативных функций (дыхания, работы сердечно-сосудистой системы);
- участие в интегративных процессах высшей нервной деятельности.
При болезни Галлервордена — Шпатца в бледном шаре и черном веществе накапливается железо, что приводит к нарушению их работы. Как следствие, возникают моторные дисфункции и психические расстройства. Впервые заболевание было описано в 1922 году медиками Юлиусом Галлерворденом и Хуго Шпатцем. Оно встречается у 3 человек из 1000000.
Причины и патогенез
Болезнь Галлервордена — Шпатца относится к наследственным патологиям: она передается по аутосомно-рецессивному принципу. Однако в медицинской литературе описаны не только семейные случаи заболевания, но и спорадические (случайные).
Причина развития недуга – мутации в PANK2-гене, расположенном в 20-й хромосоме. Он отвечает за кодирование пантотенат киназы. Данный фермент участвует в биосинтезе коэнзима А, который ускоряет фосфорилирование пантотената, пантина и N-пантотенилцистеина.
Генетический дефект приводит к снижению выработки пантотенат киназы. Как следствие, в базальных ганглиях накапливается аминокислота цистеин, обладающая способностью связывать ионы железа. В результате в мозгу формируются устойчивые соединения, провоцирующие разрушение нейрональных протеинов.
Апоптоз (гибель) нейронов усиливается под влиянием окислительных реакций, в которых участвуют свободные радикалы. В итоге разрушенные нейроны замещаются глиями – вспомогательными клетками нервной ткани.
Патологоанатомическая картина болезни Галлервордена — Шпатца характеризуется такими признаками, как:
- накапливание пигмента, содержащего железо, в бледном шаре, черной субстанции и красных ядрах, из-за чего эти зоны приобретают желтовато-коричневый оттенок;
- нейроаксональные образования (аксоны с локальными расширениями) в базальных ганглиях, коре полушарий и периферических нервах;
- гибель нейронов в разных отделах мозга;
- диффузная демиелинизация – разрушение оболочки нервных волокон.
Общий обмен железа при этом не нарушен – его количество в сыворотке крови не превышает нормы.
Симптомы
В зависимости от возраста, в котором появляются симптомы, выделяют три варианта болезни Галлервордена — Шпатца:
- инфантильный – манифестация в 1-3 года;
- типичный детский – 4-15 лет;
- поздний – 16-20 лет и старше.
Существует еще одна классификация:
- детская форма – дебют в 4-10 лет;
- ювенильная – 10-18 лет;
- взрослая (атипичная) – после 18 лет.
Патология отличается полиморфизмом симптомов. В большинстве случаев она проявляется после 5 лет. Типичные признаки детской формы:
- дистония нижних конечностей – сокращение мышц, приводящее к изменению походки;
- генерализация двигательных расстройств – постепенно в процесс вовлекаются мышцы туловища, лица, глотки, при этом наблюдаются кривошея, спазм кистей, блефароспазм и так далее;
- синдром паркинсонизма, который сопровождается гипокинезией, ригидностью мышц и тремором в состоянии покоя;
- эпилептические припадки;
- зрительные расстройства – дегенерация сетчатки, атрофия зрительных нервов;
- умственная отсталость в результате ухудшения внимания и памяти;
- агрессивность, асоциальность.
Ювенильная форма характеризуется дистонией мышц конечностей и челюстно-ротовой области, а также нейропсихологическими и поведенческими расстройствами.
Атипичный вариант болезни Галлервордена — Шпатца, дебютирующий после 18-20 лет, встречается редко – в 15% случаев. Он сопровождается такими симптомами, как:
- синдром паркинсонизма с постуральной неустойчивостью – невозможностью поддерживать равновесие;
- гиперкинезы в виде дистонии, хореоатетоза (комбинации порывистых и замедленных движений), гемибаллизма (размашистых движений с одной стороны тела), миоклонии (кратковременных мышечных спазмов);
- деменция – стойкое снижение интеллектуальных способностей;
- пирамидные знаки – патологические рефлексы, возникающие из-за поражения нейронов головного мозга.
Диагностика
Болезнь Галлервордена — Шпатца диагностируется на основании клинических симптомов, которые устанавливаются в ходе неврологического обследования. Кроме того, проводятся генетический тест, показывающий мутацию в PANK2-гене, и магнитно-резонансная томография мозга.
Позитронно-эмиссионная томография показывает снижение кровотока в стриатуме, бледном шаре и лобно-теменных долях.
Болезнь Галлервордена — Шпатца дифференцируют от болезни Вильсона-Коновалова и нейроакантоцитоза.
Лечение
Лечение болезни Галлервордена — Шпатца направлено на облегчение симптомов. Терапии, способной повлиять на причину патологии, пока не существует.
Основные медикаментозные средства:
- дефероксамин, феррипрокс – хелаторы железа, которые способны образовывать с ним химические связи и препятствовать его накоплению, но при рассматриваемом заболевании они малоэффективны;
- леводопа – противопаркинсоническое средство;
- антихолинергики для уменьшения выраженности гиперкинезов;
- баклофен, бензодиазепины, миорелаксанты и инъекции диаспора для расслабления мышц и снятия боли;
- нейромидин и глиатилин для коррекции когнитивных нарушений;
- атипичные нейролептики для лечения психических расстройств.
К новейшим методам терапии болезни Галлервордена — Шпатца относятся введение пантотеновой кислоты и магнитная стимуляция мозга.
Прогноз
Болезнь Галлервордена — Шпатца характеризуется неуклонным прогрессированием симптомов. Наиболее агрессивное течение свойственно детской форме: через 6-15 лет после проявления патологии наступает полная инвалидизация.
Поздний вариант болезни имеет более благоприятный прогноз, особенно если сопровождается маловыраженной деменцией. Благодаря терапии степень симптомов можно снизить, и сохранить способность пациента к самообслуживанию. Средняя продолжительность атипичной формы заболевания – 20 и больше лет.
Профилактика
Профилактика болезни Галлервордена — Шпатца невозможна из-за ее генетической этиологии.
МКБ-10
- Причины болезни Галлервордена-Шпатца
- Симптомы болезни Галлервордена-Шпатца
- Диагностика болезни Галлервордена-Шпатца
- Лечение и прогноз болезни Галлервордена-Шпатца
- Цены на лечение
Общие сведения
Болезнь Галлервордена-Шпатца описана в 1922 г. немецкими морфологами, в честь которых и получила свое название. К наиболее типичным клиническим маркерам данной патологии относят гиперкинезы, синдром паркинсонизма, интеллектуальное снижение, атрофию зрительных нервов, пигментную ретинопатию. Болезнь Галлервордена-Шпатца встречается крайне редко. В зависимости от времени ее манифестации различают детскую, ювенильную (подростковую) и взрослую формы. Ранее заболевание диагностировалось лишь посмертно по данным аутопсии. После внедрения в практическую неврологию МРТ стала возможна прижизненная постановка диагноза. Прорыв в изучении этиологии был сделан в 2001 г., когда было установлено, что в основе заболевания лежит генетический дефект, обуславливающий нарушения в синтезе фермента пантотенаткиназы. После этого болезнь Галлервордена-Шпатца была официально переименована в пантотенаткиназа-ассоциированную нейродегенерацию.
Причины болезни Галлервордена-Шпатца
Болезнь Галлервордена-Шпатца является генетической патологией, носящей как семейный, так и спорадический характер, передающейся по наследству аутосомно-рецессивным путем. Генетическим субстратом выступают аберрации в гене пантотенаткиназы (локус 20р12.3–р13 20-й хромосомы). Всего известно более 50 мутаций. Результатом генетического дефекта является уменьшение продукции пантотенаткиназы, что ведет к аккумуляции в базальных структурах цистеина. Последний образует устойчивые химические соединения с ионами железа, которые неблагоприятно воздействуют на белки и запускают процесс перекисного окисления, приводящий к апоптозу нейронов. На месте некротизированных нейронов происходит разрастание глиальной ткани.
Описанный патологический процесс затрагивает преимущественно бледный шар и черное вещество (субстанцию nigra), где морфологически обнаруживаются внеклеточные отложения железа, имеющие коричневую пигментацию. Кроме этого, имеют место сфероидные периаксональные образования, расположенные в белом церебральном веществе, коре мозга, спинном мозге и периферических нервных стволах.
Симптомы болезни Галлервордена-Шпатца
Классическим вариантом болезни Галлервордена-Шпатца считается ранняя детская форма с клинической манифестацией в период от 4 до 10 лет (обычно после 5-летнего возраста). В 90% случаев первым признаком заболевания выступает торсионная дистония, затрагивающая мышцы ног. Ведущей жалобой является затруднение ходьбы. Затем, как правило, происходит генерализация процесса с его распространением на мышцы глотки, лица, туловища. Наряду с генерализованными вариантами могут отмечаться мультифокальный или сегментарный тип дистонии. Наиболее часто наблюдается писчий спазм, блефароспазм, лицевой параспазм, спастическая кривошея. У трети пациентов отмечаются признаки паркинсонизма: мышечная ригидность и гипокинезия. В ряде случаев имеют место эпилептические приступы.
Болезнь Галлервордена-Шпатца характеризуется когнитивными расстройствами в виде снижения внимательности и памяти с постепенным развитием олигофрении; психическими изменениями с преобладанием агрессивности и асоциального поведения. Отмечается дизартрия. У большинства больных имеются нарушения остроты зрения. В 68% случаев они обусловлены атрофией зрительных нервов, в 29% случаев — пигментной ретинопатией. Для детской формы болезни Галлервордена-Шпатца типично быстрое прогрессирование с полной потерей в течение 10-15 лет способности к передвижению.
Подростковый вариант болезни Галлервордена-Шпатца проявляется в возрасте от 10 до 18 лет и характеризуется более замедленным течением. Дебютирует проявлениями фокальной торсионной дистонии, наиболее часто в мышцах конечностей или ортомандибулярной области. Сопровождается психическими, интеллектуальными и поведенческими расстройствами.
Диагностика болезни Галлервордена-Шпатца
Благодаря полиморфизму симптоматики, постановка диагноза болезни Галлервордена-Шпатца представляет трудную задачу для неврологов. Основными критериями заболевания считаются дебют в возрасте до 30 лет, экстрапирамидные расстройства, неуклонное прогрессирование симптомов, наличие типичной МРТ-картины. К дополнительным признакам отнесены наличие пирамидных знаков, прогрессирующее интеллектуальное снижение, эпиприступы, атрофия зрительных нервов, пигментная атрофия сетчатки, аутосомное наследование по рецессивному типу.
В диагностике опираются на данные неврологического статуса и электроэнцефалографии. При нарушении зрения проводят консультацию офтальмолога, визиометрию, офтальмоскопию. Определение типа наследования осуществляет генетик путем составления генеалогического древа. Возможна ДНК-диагностика (поиск мутаций в гене пантотенаткиназы). При проведении ПЭТ головного мозга удается выявить сниженный метаболизм в зоне паллидума. Основанием для исключения болезни Галлервордена-Шпатца является наличие симптомов другой патологии, в рамки которой может укладываться имеющаяся клиническая картина: болезни Вильсона, хореи Гентингтона, нейроакантоцитоза, болезни Мачадо-Джозефа.
Лечение и прогноз болезни Галлервордена-Шпатца
В настоящее время болезнь Галлервордена-Шпатца не имеет эффективных методов лечения. Попытки терапии препятствующими накоплению железа хелатными соединениями (дефероксамином) и антиоксидантами не имели успеха. В связи с этим применяется симптоматическое лечение. Синдром паркинсонизма служит показанием к назначению дофаминовых агонистов (пирибедила, прамипексола) или производных амантадина. Однако при данном заболевании он, как правило, резистентен к проводимому лечению.
При гиперкинезах применяют вальпроаты, бензодиазепины (диазепам, клоназепам). При спастике рекомендованы миорелаксанты (баклофен, толперизона гидрохлорид), при эпиприступах — топирамат или вальпроаты, при когнитивных расстройствах — ипидакрин и холина альфосцерат, при психических отклонениях — нейролептики (рисперидон, кветиапин, клоназепам), антидепрессанты 3-го поколения (венлафаксин, циталопрам, дапоксетин).
Симптоматическая терапия болезни Галлервордена-Шпатца позволяет уменьшить проявленность клинических симптомов, продлить способность пациентов к самообслуживанию. Вместе с тем продолжается разработка новых способов лечения. Исследуется эффективность применения пантотеновой кислоты. Получены данные о положительном влиянии на течение заболевания магнитной стимуляции бледного шара.
Прогноз зависит от формы заболевания. Наиболее неблагоприятное течение имеет ранняя форма, при которой полная инвалидизация наступает в промежутке от 10 до 15 лет с момента дебюта симптомов. Более благоприятен взрослый вариант, особенно в случаях, когда деменция слабо выражена. Его средняя продолжительность составляет более 20 лет.

В 1922 году два ученых из Германии, Юлиус Галлеворден и Гуго Шпатц, занялись изучением следующего клинического случая: в семье из 12 человек у пяти сестер наблюдались прогрессирующая деменция и дизартрия. Данные аутопсии показали, что в многочисленных областях головного мозга ткани были окрашены в коричневый цвет. В частности, интерес представляла окраска в области чёрной субстанции и базальных ядер. Описанное заболевание получило название синдрома Галлевордена — Шпатца и было под ним известно вплоть до недавнего времени, пока не была установлена роль исследователей в проведении экспериментов над заключенными в нацистской Германии, после чего эпоним сменили на нейродегенерацию с отложением железа в головном мозге (Neurodegeneration with brain iron accumulation, NBIA).
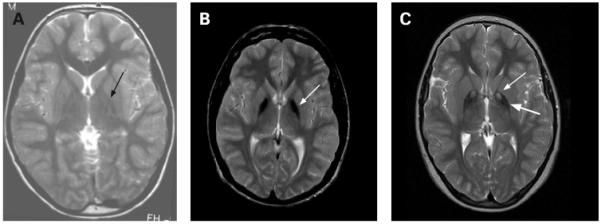
Рисунок 1. Т2-взвешенные снимки. А - здоровый пацент. В - идиопатическая NBIA, стрелкой обозначена зона гипоинтенсивности в медиальном бледном шаре. С - PKAN, регион гиперинтенсивности (тонкая стрелка) окружён гипоинтенсивной зоной (толстая стрелка), так же в медиальном бледном шаре, т.н "глаз тигра" - типичное для PKAN распределение железа.
До 2001 года синдромом Галлевордена — Шпатца называли все формы NBIA, однако после открытия первого NBIA-ассоциированного гена от этого пришлось отказаться. Изученный ген исследователи связали с пантотенат-киназно связанной нейродегенерацией (Pantothenate kinase-associated neurodegeneration, PKAN) — самой распространенной формой нейродегенерации с отложением железа. В течение последующих лет были открыты и другие генные мутации и фенотипы нейродегенерации: в 2006 году была обнаружена мутация гена PLA2G6, кодирующего фосфолипазу А2, приводящая к возникновению PLA2G6-ассоциированной нейродегенерации (PLAN), в 2011 — ген C19orf12 был идентифицирован как ответственный за нейродегенерацию, ассоциированную с белками митохондриальной мембраны (MPAN). Еще более недавно в спектр NBIA включили еще одно заболевание: бета-пропеллерный белок-ассоциированную нейродегенерацию (BPAN). Вместе эти четыре подтипа нейродегенерации с отложением железа являются наиболее часто встречающимися и диагностируемыми (более чем 62 % от всех случаев).
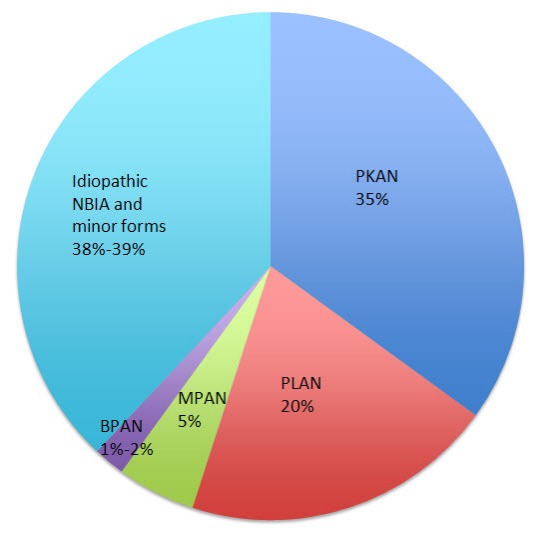
Рисунок 2. Частота встречаемости подтипов NBIA.
В спектр также были включены еще 6 более редких подтипов: нейроферритинопатия, ацерулоплазминемия, болезнь Куфора — Ракеба, нейродегенерация, связанная с гидроксилазой жирных кислот (FAHN), синдром Вудхауза — Сакати и COASY-протеин-ассоциированная нейродегенерация (CoPAN). Большая часть заболеваний, объединенных термином NBIA наследуется аутосомно-рецессивно, за исключением BPAN и нейроферритинопатии, которые наследуются Х-сцепленно доминантно и аутосомно-доминантно соответственно.
В случаях, когда этиология заболевания не может быть установлена, пациенту диагностируют идиопатическую нейродегенерацию с накоплением железа. Особенно усложняется постановка диагноза в том случае, если пациент — единственный в семье человек с заболеванием. Даже если есть вероятность наследования болезни по аутосомно-рецессивному типу, это следует подтвердить при помощи проведения молекулярно-генетического исследования.
Течение заболевания очень сильно варьирует: манифест может случиться в любом возрасте, начиная от младенчества. Прогрессируют симптомы в ряде случаев стремительно, а в ряде — медленно, с длительными периодами стабильности. Конкретно симптоматика определяется генетическими механизмами, вызвавшими нейродегенерацию, но факторы, влияющие на тяжесть и скорость прогрессирования все еще остаются неизвестны.
Характерными признаками NBIA являются прогрессирующая дистония, дизартрия, спастичность, паркинсонизм и отставание в интеллектуальном развитии. Часто симптомы сопровождаются дегенерацией сетчатки и атрофией зрительного нерва. Среди нейропатологических признаков отмечают не только накопление железа, но еще и образование аксональных сфероидов в ЦНС и иногда в периферических нервах.
Существует корреляция между скоростью прогрессирования болезни и возрастом появления первых симпотомов. Несмотря на это, были зарегистрированы случаи очень быстрого ухудшения состояния у лиц с манифестом в раннем подростковом возрасте вплоть до смерти до достижения 20 лет. Напротив, есть и пациенты, дожившие до 50 лет, несмотря на проявление первых симптомов в возрасте до 10.
По мере ухудшения состояния пациентам часто требуется кормление через гастральный зонд из-за развития дисфагии. Гастроэзофагеальный рефлюкс и запоры становятся хронической проблемой для больных на поздних стадиях заболевания. Обычно причиной смерти становятся вторичные осложнения, например, аспирационная пневмония и истощение.
Исторически, NBIA ассоциировалась исследователями с нарушениями интеллекта. В исследовании с 16 пациентами с PKAN оба из проведенных тестирований выявили отрицательную корреляцию уровня IQ с тяжестью заболевания. То есть испытуемые с самыми тяжелыми проявлениями хуже всего справились с тестами.
В случае если речь идет о других формах заболевания из спектра, постановка диагноза может оказаться гораздо более затруднительной, чем при PKAN. В этом случае на помощь приходит МРТ, подходящая для первичного определения формы нейродегенерации, тем самым наталкивая врача на выбор правильного молекулярно-генетического исследования. МРТ головного мозга является стандартным методом исследования для описываемой группы заболеваний. Более новые нейровизуализационные технологии, например, магнитно-резонансная спектроскопия (МРС), предположительно, тоже могут оказаться полезными для постановки диагноза, однако на данный момент это достоверно неизвестно.
По определению в базальных ганглиях людей с NBIA определяется ненормально высокое количество железа. Обычно эти зоны определяются как гипоинтенсивные участки бледного шара и ретикулярной части черной субстанции на Т2-взвешенных снимках. На Т1-взвешенных снимках эти области изоинтенсивны, что помогает отличить их от отложений кальция и прочих изменений. При идиопатической нейродегенерации с накоплением железа участки его аккумуляции могут встречаться в красном ядре, зубчатом ядре, скорлупе и хвостатом ядре. После постановки диагноза проводить повторные исследования при помощи МРТ не имеет смысла. Следует помнить, что порой диагностика с целью выявления конкретной формы NBIA может занять несколько лет. Например, в случае болезни Куфора — Ракеба или PLAN накопление железа может произойти только в ряде случаев.
Как фармакологические, так и хирургические вмешательства могут быть направлены только на паллиативную терапию в связи с этиологией нейродегенераций. Многие из этих вмешательств предоставляют лишь временное улучшение, поэтому для поддержания максимального высокого качества жизни пациента необходимы периодические корректировки хода лечения. Наиболее эффективными препаратами для снятия дистонии и спастичности являются баклофен и тригексифенидил. Как правило, пациенты с PKAN не реагируют на L-дофа, в то время, как пациенты с другими формами NBIA и паркинсонизмом в некоторых случаях реагируют на L-дофа, особенно в редких случаях, характеризующихся ранними задержками развития и проявлением дистонии уже во взрослом возрасте. Ботулотоксин может быть полезен для пациентов, чье качество жизни может быть улучшено терапией конкретной части тела. Например, инъекции в мышцы лица способствуют улучшению речи и способности к самостоятельному питанию.
Читайте также: