
Арнольда киари шейного отдела

Синдром Арнольда-Киари — врожденная патология краниовертебральной зоны, возникающая в период эмбриогенеза и характеризующаяся деформацией черепа. Опущение структур головного мозга в большое затылочное отверстие приводит к их сдавлению и ущемлению. Дисфункция мозжечка сопровождается нистагмом, шаткой походкой, дискоординацией движений, а поражение продолговатого мозга — изменениями в работе жизненно важных органов и систем.
Впервые синдром был описан в конце 18 века двумя учеными: доктором из Германии Арнольдом Джулиусом и врачом из Австрии Гансом Киари. Аномалию обнаруживают сразу после появления ребенка на свет или несколько позже — в пубертатном или зрелом возрасте. Это зависит от типа синдрома. У взрослых пациентов недуг чаще всего становится неожиданной находкой. Средний возраст больных — 25-40 лет.
Симптоматика патологии также определяется типом мальформации. Хоть синдром и считается врожденным пороком, не всегда его клинические признаки возникают с самого рождения. Иногда они обнаруживаются после 40 лет. У больных возникает цефалгия, напряженность мышц шеи, головокружение, нистагм, обмороки, атаксия, нарушения речи, парез гортани, тугоухость, падение остроты зрения, нарушение процесса глотания, стридор, парестезии, слабость мышц. У большинства пациентов симптомы заболевания полностью отсутствуют. Его обнаруживают случайно, во время комплексного диагностического обследования, проводимого совсем по другому поводу. Пациентам с бессимптомными формами не требуется лечение.
При постановке диагноза учитывают данные осмотра больного и его неврологического статуса, а также результаты томографического исследования мозга. Магнитно-ядерный резонанс позволяет точно и быстро определить наличие трансформации, уровень поражения и степень ущемления головного мозга. Лечение синдрома медикаментозное, физиотерапевтическое и оперативное. Больным выполняют операции по шунтированию мозга и декомпрессии краниовертебральной зоны.
Этиология
Причины синдрома в настоящее время точно не определены. Существует несколько теорий происхождения патологии, но до конца ни одна из них не имеет официального подтверждения. Мнения ученых-неврологов всего мира до сих пор расходятся.
Большинство медиков признают синдром врожденным недугом, сформированным в процессе эмбриогенеза под воздействием негативных факторов внешней среды, оказывающих свое пагубное действие на женский организм при беременности. К ним относятся: самостоятельные использование лекарств, алкоголизм, табакокурение, вирусные инфекции, ионизирующее излучение.

- Дистопия мозжечковых миндалин за пределы задней черепной ямки, имеющей относительно малые размеры;
- Выталкивание растущих структур мозга через затылочное отверстие;
- Неправильное формирование в процессе эмбриогенеза и атипичное развитие в постнатальном периоде костной ткани, приводящее к деформации черепной коробки.
Некоторые ученые определенную роль в развитии синдрома отводят генетическому фактору. В настоящее время можно точно утверждать, что болезнь не связана с хромосомными аномалиями.
Другие ученые придерживаются иной точки зрения относительно происхождения синдрома. Они считают его приобретенным и объясняют свое мнение появлением симптомов патологии у взрослых лиц. Приобретенный синдром возникает под действием экзогенных факторов. У больного новорожденного ребенка череп может иметь нормальное строение без костных аномалий и гипоплазии.
- Родовой травматизм с поражением черепа и мозга,
- Воздействие цереброспинальной жидкости на стенки спинного мозга,
- Любые ЧМТ,
- Бурный рост мозга в условиях медленно растущих костей черепа.
Симптомы патологии долгое время отсутствуют у больных, а затем внезапно появляются под воздействием провоцирующих факторов: вирусов, травм головы, стрессов.
Опущение основных мозговых структур до шейных позвонков блокирует процесс перетекания ликвора из подпаутинного пространства в спинномозговой канал. Это приводит к дисциркуляторным изменениям. Ликвор, продолжая синтезироваться и никуда не оттекая, скапливается в головном мозге.
Ученый Киари в 1891 году выделил четыре типа аномалии:
- I – выход структурных элементов головного мозга за пределы задней черепной ямки, обусловленный недоразвитием костной ткани этой области. Этот тип клинически проявляется у лиц зрелого возраста.

выход структур мозжечка за пределы ЗЧЯ при аномалия 1 типа
- II — нарушения эмбриогенеза, приводящие к расположению структур мозжечка и продолговатого мозга ниже большого затылочного отверстия.

синдром Арнольда-Киари II типа
- III — эктопия мозговых структур в каудальном направлении с образованием энцефаломенингоцеле.

аномалия III типа
- IV — недоразвитый мозжечок не смещается и не выходит за пределы черепа. Поскольку отсутствует грыжевое выпячивание мозга, этот тип синдрома отсутствует в современной классификации.
Существуют два новых типа синдрома. Тип 0 — мозжечок располагается достаточно низко, но при этом находится в черепной коробке. Тип 1.5 – промежуточная форма, сочетающая признаки I и II типов.
Выделяют три степени тяжести патологии:
- Первая — относительно легкая форма патологии без аномалий мозговых структур и характерных клинических проявлений.
- Вторая — наличие пороков развития ЦНС с врожденным недоразвитием головного мозга и подкорки.
- Третья — аномалии строения головного мозга со смещением мягких тканей в отношении твердых структур, образованием ликворных кист и сглаженностью извилин.
Симптоматика

Аномалия Киари I типа — самая распространенная форма синдрома, клинические признаки которой условно объединены в пять синдромов:
- Гипертензионный синдром проявляется цефалгией, подъемом артериального давления в утренние часы, напряженностью и гипертонусом шейных мышц, дискомфортом и болезненными ощущениями в шейном отделе позвоночника, диспепсическими явлениями, общей астенизацией организма У новорожденных детей возникает общее беспокойство, рвота фонтаном, тремор подбородка и конечностей, нарушается сон. Ребенок постоянно плачет, отказывается от груди.
- При наличии мозжечковых нарушений у больных изменяется произношение, речь становится скандированной, возникает вертикальный нистагм. Они жалуются на частые головокружения, рассогласованность движений, шаткость походки, дрожание рук, нарушение равновесия, дезориентацию в пространстве. Больные с большим трудом выполняют простые целенаправленные действия, в движениях отсутствует четкость и скоординированность.
- Поражение черепно-мозговых нервов проявляется признаками корешкового синдрома. У пациентов ограничивается подвижность языка и мягкого неба, что приводит к нарушению речи и проглатывания пищи. Их голос изменяется в сторону гнусавости и осиплости, речь становится неясной, дыхание затрудненным. Нарушение ночного дыхания отмечаются у большинства больных. У них возникает гипопноэ, центральное или обструктивное апное, при прогрессировании которого развивается острая дыхательная недостаточность. Лица с синдромом плохо слышат и видят, у них двоится в глазах и шумит в ушах. Со стороны органов зрения пациенты отмечают наличие светобоязни и боль при движении глазными яблоками. Офтальмологи часто обнаруживают анизокорию, спазм аккомодации или скотомы. Одним из основных симптомов синдрома является гипестезия – снижение чувствительности кожи лица и конечностей. Подобные патологическое изменения связаны с приглушенным реагированием рецепторов кожи на внешние раздражители: тепло или холод, уколы, удары. В тяжелых случаях нервное окончания вообще перестают воспринимать различные экзогенные воздействия.
- Сирингомиелический синдром — сложный симптомокомплекс, проявляющийся парестезией или онемением конечностей; изменением тонуса мышц и их гипотрофией, приводящей к миастеническим расстройствам; поражением периферических нервов, проявляющимся болью в конечностях; дисфункцией органов таза в виде затрудненной дефекации или самопроизвольного мочеиспускания; возможны артропатии — поражения суставов.
- У больных с пирамидальной недостаточностью снижается сила в нижних конечностях и способность к тонким движениям, ограничивается объем движений, повышается мышечный тонус — так называемая спастичность, например, спастическая походка. Повышение сухожильных рефлексов сочетается с одновременным снижением кожных рефлексов — брюшных. Возможно появление патологических рефлексов. У пациентов страдает мелкая моторика рук.
Любое неосторожное движение усиливает симптомы патологии, делает их более выраженными и яркими. Изменение положения головы — частая причина потери сознания.
Синдром Киари II типа имеет сходные клинические проявления. У новорожденных возникает паралич гортани, врожденный стридор, ночное апноэ, дисфагия, срыгивания, нистагм, гипертонус мышц рук, цианоз кожи. Аномалии III и IV типов не совместимы с жизнью.
Диагностические мероприятия

Аномалия Арнольда-Киари на снимке МРТ
Врачи-неврологи и невропатологи осматривают пациента и выявляют характерные особенности походки, изменение рефлексов и чувствительности на определенных участках тела, слабость в руках и прочие признаки. Все проявления мозжечкового, гидроцефального, бульбарного и прочих синдромов в совокупности позволяют врачу заподозрить аномалию.
После определения неврологического статуса больного требуется проведение комплексного неврологического обследования, включающего инструментальные методы — электроэнцефалографию, УЗИ головного мозга, реоэнцефалографию, ангиографию, рентгенографию. Эти методики выявляют лишь косвенные признаки патологии — изменения, происходящие в организме больного.
Ядерный магнитный резонанс лежит в основе особого нерентгенологического метода исследования – томографии. Этот спектроскопический анализ безопасен для большинства людей. Он дает изображение, состоящее из тонких срезов от магнитнорезонансного сигнала, проходящего через тело человека. На сегодняшний день именно МРТ позволяет быстро и точно поставить диагноз. Томография визуализирует структуру костей и мягкие ткани черепа, определяет пороки мозга и его сосудов.
Лечебный процесс
Лечение аномалии Киари комплексное, включающее медикаментозное воздействие, физиотерапевтические процедуры и хирургическое вмешательство. Именно оно в большинстве случаев помогает справиться с недугом и восстановить нормальную работу всего организма. Возможно применение средств народной медицины, которые дополняют, но не заменяют основное лечение. Использование фитосборов, отваров и настоев лекарственных трав должно быть одобрено лечащим врачом.
Если больные испытывают сильную головную боль, боль в шее, мышцах и суставах, им назначают следующие группы препаратов:
Патогенетическое лечение синдрома включает:

Если состояние больного признают крайне тяжелым, то его госпитализируют сразу в реанимационное отделение. Там пациента подключают к аппарату ИВЛ, устраняют имеющийся отек мозга, предупреждают инфекционные патологии и корректируют неврологические нарушения.
Физиотерапевтическое воздействие дополняет медикаментозное лечение, позволяет быстрее добиться положительных результатов, ускоряет процессы восстановления функций организма и выздоровления больных. Неврологи назначают:
- Криотерапию, оказывающую обезболивающий эффект, стимулирующую работоспособность желез внутренней секреции и укрепляющую иммунитет.
- Лечение лазером, улучшающее трофику и микроциркуляцию в очаге поражения.
- Магнитотерапию, оказывающую общее оздоравливающее действие и запускающую внутренние резервы организма.
В настоящее время особой популярностью пользуется кинезиологическая терапия, которая направлена на развитие умственных способностей и достижение физического здоровья через двигательные упражнения. Ее также включают в схему лечения данного синдрома.
Лечение не проводится вообще, если патология была обнаружена случайно, во время прохождения томографического обследования совсем по другому поводу, и у больного отсутствуют какие-либо характерные симптомы. За состоянием таких пациентов специалисты ведут динамическое наблюдение.
Стойкие неврологические нарушения с парестезиями, дистонией мышц, параличами и парезами требуют проведения хирургической коррекции. Оперативное вмешательство показано также в тех случаях, когда медикаментозная терапия на дает положительного результата. Операции преследуют одну цель — устранение сдавления и ущемления мозга, а также восстановление нормальной циркуляции ликвора.

В настоящее время нейрохирурги спасают жизнь больным путем выполнения декомпрессивных и шунтирующих операций. В первом случае выпиливают часть затылочной кости с целью расширения большого отверстия, а во втором создают обходной путь для оттока ликвора по имплантационным трубкам с целью снижения его объема и нормализации внутричерепного давления.
После хирургического вмешательства всем пациентам показаны реабилитационные мероприятия. Если лечение было успешным, у больных восстанавливаются утраченные функции — дыхательные, двигательные, сердечно-сосудистые, нервные. В течение трех лет возможно рецидивирование патологии. В таких случаях больных признают инвалидами.
Народные средства, применяемые при данной патологии, устраняют боль и расслабляют напряженные мышцы. Они эффективно дополняют традиционную терапию синдрома.
Наиболее популярные средства:
- Настой алтея для постановки компрессов,
- Прогревания пораженного места горячим куриным яйцом,
- Медовые компрессы,
- отвар папоротника или малины для приема внутрь.
Синдром Арнольда-Киари – порок развития, протекающий в бессимптомной форме или проявляющийся клинически с момента рождения. Патология имеет весьма разнообразную симптоматику и подтверждается с помощью МРТ. Лечебный подход к каждому пациенту индивидуален. Тактика лечения варьируется от симптоматического воздействия медикаментами до оперативного вмешательства с трепанацией черепа и удалением части мозговых структур.
Профилактика
Поскольку этиология синдрома окончательно не выяснена и нет конкретной информации о его патогенезе, предупредить развитие патологии не представляется возможным. Будущим родителям необходимо знать все о ведении здорового образа жизни и при планировании беременности стараться соблюдать указанные правила:
- Отказаться от пагубных привычек в виде табакокурения и употребления спиртных напитков,
- Обогащать свой рацион белковыми продуктами, фруктами, овощами, ягодами, исключив из него сладости и вредности,
- Своевременно обращаться к врачам за медицинской помощью,
- Принимать лекарственных препараты по назначению врача и в строго указанных дозировках,
- С профилактической целью принимать поливитамины,
- Беречь свое здоровье и наслаждаться жизнью.
Прогноз патологии неоднозначный. Консервативное лечение часто не дает положительных результатов. Хирургическое вмешательство, выполненное своевременно и в полном объеме, не всегда восстанавливает утраченные функции организма. Согласно статистическим данным эффективность такого лечения редко превышает 50-60%. Синдром третьей степени имеет неблагоприятный прогноз, поскольку поражаются многие мозговые структуры. При этом возникают несовместимые с жизнью функциональные нарушения.
Видео: лекция по синдрому Арнольда-Киали
Общие сведения
Синдром Арнольда-Киари является пороком развития мозжечка – отдела головного мозга, отвечающего за координацию, мышечный тонус и равновесие. Патологии присвоен код по МКБ-10 Q07.0 и она представляет собой опущение миндалин мозжечка вниз на уровень первого, а порой второго шейного позвонка (ниже черты Чемберлена) и блокирует нормальный ток спинномозговой жидкости.
Заболевание чаще всего сочетается с микрогирией, сдавливанием заднего отдела мозга, стенозом водопровода мозга, базилярной импрессией, инвагинацией, недоразвитием четверохолмия и другими мальформациями нервной системы. Синдром чаще всего встречается у особ в возрасте 12-71 год и не превышает 000,9%.
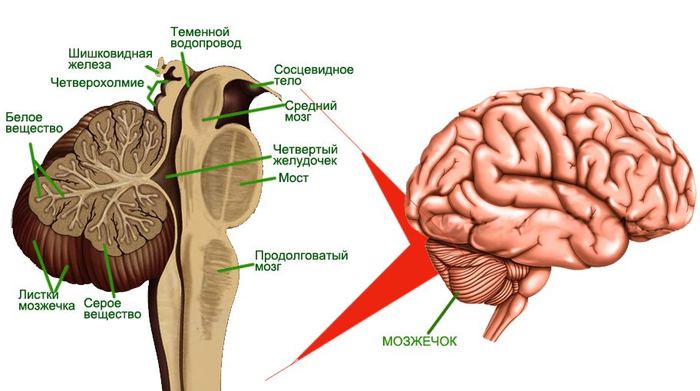
Локализация и строение мозжечка
Патогенез
В основе патофизиологии обычно лежит несоответствие размеров задней черепной ямки и имеющихся в ней структур нервной системы, а также:
- развитие аномалий тел шейных позвонков, включая их расщепление чаще всего первого (данный механизм развития встречается в 5% случаев), ассимиляцию атланта — сращивание шейного позвонка с затылочной костью;
- смещение структур мозжечка в период бурного роста мозга при медленно растущих костях черепа;
- гидроцефалия – избыточное скопление цереброспинальной жидкости;
- сирингомиелия – анормальный процесс развития полостей в спинном мозге;
- миеломенингоцеле – врожденный дефект развития нервной трубки;
- различные врожденные заболевания, в том числе платибазия, аномалия Денди-Уокера.
Классификация
В зависимости от клинической картины и степени развития анатомических аномалий синдром Арнольда-Киари бывает четырех типов.
Синдром Арнольда-Киари 1 типа проявляется в виде проникновения миндалин мозжечка в полость позвоночного канала, вызывающего гидромиелию и опущение структур задней черепной ямки ниже большого затылочного отверстия на 3-5 мм и более, причем нет никаких других мальформаций нервной системы. Средняя продолжительность жизни обычно не превышает 25-40 лет.
Представляет собой опущение в полость позвоночного канала различных структур мозжечка и тканей ствола, при этом данная нейропозвоночная мальформация сочетается с миеломенингоцеле (врожденной спинномозговой грыжей) и гидроцефалией. Манифестация происходит практические сразу после рождения.
Мальформация отличается наличием затылочного энцефалоцеле и различных признаков аномалии второго типа. Обычно не совместима с жизнью.
По своей сути это аплазия либо гипоплазия всех или отдельных структур мозжечка, то есть бывает тотальной и субтотальной. Первый вариант встречается достаточно редко и сочетается с прочими тяжёлыми аномалиями и заболеваниями нервной системы, включая анэнцефалию, амиелию. При субтотальной агенезии наблюдаются пороки развития других участков головного мозга, например агенезия моста, отсутствие четвёртого желудочка и пр.
Гипоплазия мозжечка встречается в форме уменьшения всего мозжечка или охватывает отдельные части, при этом сохраняются нормальные структуры без утраты функций. Встречается одно- и двусторонняя, лобарная, лобулярная и интракортикальная гипоплазия. Изменения конфигурации листков мозжечка обычно представлено в виде аллогирии, полигирии или агирии.
Кроме того, некоторые авторы выделяют два дополнительных типа:
Причины
Помимо роли наследственного и генетического фактора существует несколько теорий возникновения пороков мозжечка. Традиционная теория говорит, что опущение миндалин вызвано натяжением струны спинного мозга в результате напряжения концевой нити при развитии той или иной мальформации. Исключением становится болезнь Киари 1 типа, ведь единственным нарушением в этом случае становится опущение миндалин и оно может быть спровоцировано:
- гидродинамическими явлениями — нарушением циркуляции спинно-мозговых жидкостей;
- черепно-мозговыми и родовыми травмами;
- мальформацией — маленькие размеры и ограниченность затылочного отверстия могут приводить к опущению миндалин в просвет позвоночного канала;
- анормально натянутой связкой — так называемой концевой нитью (по теории доктора М.Б. Ройо Сальвадора — Filum System).
Кроме того, ученые выделяют ряд факторов, которые могут повысить риск развития аномалии Арнольда-Киари первого типа:
- генетическая предрасположенность – наличие патологии у родителей и более дальних предков, хотя хромосомных аномалий до сих пор не выявлено;
- травмы, особенно падения, могут вызвать компрессию и усиление натяжения концевой нити и привести к опущению миндалин мозжечка;
- неправильное поведение и вредные привычки женщины в период вынашивания младенца — злоупотребление и хаотичный прием медикаментов, курение, употребление алкоголя, а также перенесенные вирусные заболевания.
Симптомы
Симптоматика при различных типах синдрома Арнольда Киари может существенно отличаться – все зависит от степени натяжения и компрессии нервных структур затылочного отдела, но чаще всего у больных наблюдаются:
- головные боли;
- прогрессирующее увеличение размеров окружности головы;
- периодические боли в различных отделах позвоночника;
- парез и боль в различных областях конечностей;
- нарушения зрения и чувствительности, включая дизестезии и парестезии;
- шумы в ушах;
- паралич лицевого, глазодвигательного нерва;
- дрожь;
- приступы головокружения;
- бессонница;
- рвота;
- обмороки;
- нистагм;
- апноэ;
- нарушение работы сфинктеров, мышц языка, глотки, онемение и различные проявления мышечной слабости;
- нарушения способности глотания (дисфагия);
- нарушения памяти;
- несогласованность движений (атаксия) и как следствие — неуклюжая походка, причем нарушения наиболее ярко выражены на этапе формирования;
- сколиоз;
- трудности с удержанием позиций тела и равновесия, а также координации;
- затруднения при желании выразить мысль и подобрать слова.
Проявления синдрома Арнольда-Киари хронические, имеют тенденцию увеличивать интенсивность, что с каждым разом существенно ухудшает состояние больного и ограничивает его привычный образ жизни.
Самое опасное, что аномалия Арнольда-Киари может привести к внезапной смерти, ведь спинно-мозговые центры отвечают за сердечно-дыхательные функции, а давление миндалин мозжечка на них может спровоцировать остановку дыхания — апноэ, которое станет причиной летального исхода.
Патология 1 степени обычно выявляется случайно во время проведения МРТ, ведь больные помимо эпизодов апноэ и обмороков могут испытывать только:
- боль в шейно-затылочном отделе;
- снижение чувствительности.
Анализы и диагностика
При первых признаках необходимо пройти неврологический осмотр и провести оценку выраженности клинико-функциональных нарушений. Для подтверждения диагноза чаще всего применяются:
- нейровизуализационной методики, наиболее предпочтительно МРТ;
- электроэнцефалография;
- рентгенография черепа;
- миелография, которая позволяет выявить дефекты верхнего шейного отдела спинного мозга, ствола мозга, а также локализацию мозжечка ниже черты Чемберлена в области затылочного отверстия.
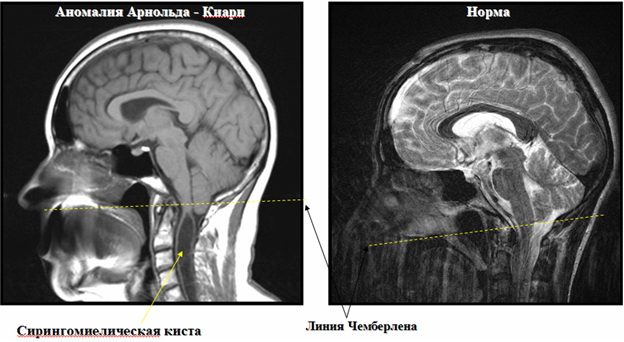
Результаты МРТ при аномалии Арнольда Киари с сирингомиелической кистой и в норме
Лечение
Тактика лечения при различных типах пороков развития мозжечка Арнольда-Киари обычно является консервативной либо продумывается нейрохирургом и представляет собой декомпрессию, наложение шунта (при выраженной гидроцефалии) или краниотомию затылочного отверстия.
Однако, благодаря докторской диссертации доктора М.Б. Ройо Сальвадора была разработана новаторская техника этиологического лечения — Filum System, которая направлена на устранение причины заболевания и патологического механизма натяжения путем хирургического минимально инвазивного рассечения концевой нити. Преимуществом методики является возможность остановить болезнь Арнольда Киари при минимальных рисках (смертность – 0%), главное выявить мальформацию и провести операцию как можно раньше. Она обычно занимает не более 45 минут и позволяет добиться симптоматического улучшения состояния, а в отдельных случаях даже поднятия миндалин мозжечка. Несмотря на короткий восстановительный период, методика имеет ряд недостатков:
- после операции остается небольшой шов;
- может возникать субъективное ощущение снижения силы конечностей;
- улучшение мозгового кровообращения вначале постоперативного периода может вызвать перепады настроения.
Но это незначительные неудобства по сравнению с минусами затылочной краниотомии, у которой:
- смертность 1-12%;
- причина заболевания не устраняется, поэтому улучшения сохраняются непродолжительный период;
- последствия оперативного вмешательства могут быть очень серьёзными и непредсказуемыми, включая отек мозга, дальнейшее опущение миндалин мозжечка, усугубление неврологической симптоматики, гемодинамические нарушения, гидроцефалию, пневмоэнцефалию, внутримозговые кровоизлияния, тетрапарез, неврологический дефицит и т.д.

Аномалия Арнольда-Киари — это отклонение в развитии. И конкретно отклонение касается связки головного и спинного мозга. Головной мозг переходит в спинной на уровне большого затылочного отверстия. Четкой границы перехода нет.
Однако расположение ствола мозга относительно костей черепа и шейного отдела позвоночника по разным причинам может меняться. Это несовпадение приводит к сжатию спинного мозга в районе шейного отдела. Нормальная циркуляция спинномозговой жидкости нарушается.
Эта ситуация еще называется мальформация. Название происходит от латинских malus (что означает плохой) и formation (образование). Проще говоря возникает аномалия развития, за которой стоят изменение строения и функций.
В данном случае имеет место мальформация Арнольда-Киари в месте перехода черепа в позвоночник, то есть мальформация головного мозга.
Описали эту патологию австрийский патологоанатом Ханс Киари (в 1891 году) и немецкий патологоанатом Юлиус Арнольд (в 1894 году). Отсюда и сложное название.
Статистика указывает на то, что частота заболевания не такая уж и редкая — до 8.4 заболевших на 100 тысяч населения. Дополнительно к аномалии (до 80% случаев) выявляется сирингомиелия (образование кист в ткани спинного мозга).
Аномалия Арнольда-Киари — что это
Справочно. Аномалия Арнольда-Киари — это нарушение развития, которое приводит к опущению мозжечковых миндалин ниже анатомического уровня и к ущемлению продолговатого мозга.
Нормой считается расположение миндалин мозжечка выше большого затылочного отверстия. В процессе мальформации они могут сместиться даже до уровня второго шейного позвонка. При таком смещении усиливается блокировка тока спинномозговой жидкости.
Не всегда удается распознать это заболевание сразу, а затем происходит его резкая манифестация. Проявление патологии происходит к 25 — 40 годам.
При обнаружении характерных для аномалии симптомов необходимо обратиться к врачу, в противном случае риск развития инфаркта спинного мозга значительно возрастает.
Поскольку речь идет об отклонении от нормального развития организма, заболевание часто называют мальформация Арнольда-Киари.
Особенности мальформации
Справочно. Задний отдел мозга перемещается к большому затылочному отверстию (БЗО), когда у черепной ямки оказываются слишком малые размеры. В итоге формируется порок, возникают нарушения циркуляции ликвора.
Кости черепа не позволяют БЗО менять свой диаметр, поэтому при любых смещениях мозговых структур ущемляются близлежащие ткани. Последствия такого явления могут носить для пациента фатальный характер.
Продолговатый мозг отвечает за работу сердечно-сосудистой и дыхательной системы организма. Его сдавливание приводит не только к неврологическому дефициту, но может давать более опасные последствия, вплоть до смерти больного.
Смещение правого и левого полушарий мозжечка останавливает движение ликвора, что провоцирует гидроцефалию. Водянка повышает риск осложнений тех расстройств, которые уже имеются у пациента.
Процент пациентов с врожденной формой аномалии Киари невелик.
Справочно. Последние данные говорят о приобретенном характере. Дистопия миндалин мозжечка обусловлена быстрым ростом тканей при медленно протекающих изменениях в строении черепа. В медицине также известна под названием эктопия миндалин мозжечка.
Синдром Арнольда-Киари без проявления симптоматики может случайно обнаружиться во время проведения МРТ.
Причины развития аномалии
Мнения медиков о причинах появления аномалии расходятся. Есть несколько теорий, объясняющих то, каким образом развивается порок.
Неврологи выделяют две патологии, приводящие к формированию мальформации Киари (Chiari malformation):
- Развитие плода в утробе матери может пойти с нарушением — черепная ямка окажется меньше анатомической нормы, отделы мозга приобретут обычные параметры.
- Размеры отделов увеличены, при этом параметры задней черепной ямки и БЗО отвечают нормам. Увеличивающийся мозг устремляется к отверстию.
Врожденная аномалия у ребенка развивается, если беременная женщина не контролирует прием лекарств, употребляет алкоголь, курит на ранних сроках беременности. 
Кроме того, вирусные инфекции у будущей матери (краснуха, цитомегаловирус) могут пагубно сказаться на развитии плода.
Справочно. Возникновению заболевания могут предшествовать различные родовые травмы, гидроцефалия, сильные повреждения головы у взрослого человека.
Типы аномалии
Рассматриваются четыре типа. Классификация производится с ориентацией на определенные основания.
Существенными признаками оказываются следующие изменения: те, что произошли в головном мозге на структурном уровне, те, что говорят о недоразвитости черепной коробки.
Аномалия Арнольда-Киари 1 типа отличается сдвигом мозжечковых миндалин, сопровождается нарушением циркуляции ликвора.
Последний заполняет узкий канал спинного мозга, вызывая гидромиелию. Указанный тип аномалии носит благоприятный прогноз. Он часто диагностируется у подростковой и взрослой групп населения.
Аномалия Арнольда-Киари 2 типа проявляет себя у новорожденных детей. Здесь наблюдается еще большее смещение отделов. Помимо мальформации, у грудничков диагностируется спинномозговая грыжа, обнаруживается аномальное развитие позвоночного столба.
В области затылка происходит выпячивание мозгового вещества через мягкую оболочку, мозжечок оказывается там же. Такова картина аномалии Арнольда-Киари 3 типа.
Внимание. Аномалия Арнольда-Киари 4 типа дает о себе знать тем, что мозжечок новорожденного оказывается недоразвитым, не занимает должного анатомического положения. Такая патология делает младенца неприспособленным к жизни, летальный исход неизбежен.
Степени тяжести
Сколько живут с аномалией Арнольда-Киари 1-й степени? Такой вопрос часто задают люди, услышавшие свой диагноз. Такая степень тяжести является самой невысокой, клинические проявления могут не отмечаться.
Спровоцировать возникновение симптомов могут черепно-мозговые травмы и повреждения позвоночника в верхней его части. Также запустить процесс может развившаяся нейроинфекция.
Аномалии 2 и 3-й степени уже сопровождаются патологическими изменениями в нервной ткани. У больного часто обнаруживают:
- смещение мозгового вещества;
- кисты проводящих ликвор путей;
- недоразвитость некоторых извилин мозга;
- гипоплазию подкорковых узлов.
Аномалия Арнольда-Киари — симптомы
Говоря о симптомах аномалии, надо, прежде всего, различать вариационные ее типы. Первый тип мальформации сопровождается несколькими синдромами, среди которых: гипертензионный, церебеллярный, сирингомиелический, бульбарный и т.д.
Гипертензионный синдром представляет собой повышение давления внутри черепа (ВЧД). Характерными симптомами будут интенсивные затылочные боли, тошнота, рвота, ригидность шейных мышц.
Церебеллярный синдром характеризуется речевыми расстройствами, нарушениями двигательной функции. При этом отсутствует четкость движений, затруднена мелкая моторика.
Сирингомиелический синдром проявляется потерей чувствительности в конечностях. Больной может получить случайный ожог, не заметив этого сразу. При обследовании обнаруживаются кисты спинного мозга.
Другие типы аномалии сопровождаются более тяжелой симптоматикой.
Справочно. У новорожденных страдает дыхание, возможна его остановка, отмечаются нарушения в глотании. Ребенок не может полноценно питаться. Посинение кожных покровов, гипертонус мышц, нистагм — вот основные проявления аномалии этих типов.
Возможные осложнения
Мальформация в некоторых случаях провоцирует достаточно опасные осложнения и может привести многих пациентов к инвалидности. Часто отмечаются увеличение ВЧД, дыхательные расстройства, апноэ, на фоне мальформации развиваются инфекционные заболевания легких и мочеполовой системы.
Справочно. Тяжело протекающая патология может стать причиной наступления комы, остановки в работе сердца, в итоге, быстрой смерти.
В запущенных случаях реанимация позволяет лишь поддерживать жизненно важные функции, сдавленный мозг восстановить практически невозможно.
Постановка диагноза
Осмотр невролога, сбор анамнеза представляют собой часть диагностики — они необходимы, но недостаточны.
Энцефалограмма, диагностика нарушения кровообращения в головном мозге и шейном отделе позвоночника могут косвенно показать наличие ВЧД. 
С помощью рентгенографии, компьютерной томографии можно зафиксировать дефекты формирования черепной коробки. Но для определения состояния нервной ткани такие способы окажутся малоинформативными.
Справочно. МРТ сегодня считается единственно достоверным методом, позволяющим провести точную диагностику и своевременно распознать синдром Арнольда-Киари.
Процедура предполагает полное обездвиживание пациента, чего легко добиться от взрослого человека. Трудности возникают, когда пациентами являются маленькие дети. В этом случае необходимо применение общего наркоза.
Варианты лечения
После постановки диагноза лечение больного осуществляет нейрохирург или невролог. В исключительных случаях устранение аномалии возможно лишь путем проведения операции.
Если единственный симптом болезни — головная боль, то врачи ограничиваются медикаментозной терапией. Специалисты подбирают препараты:
- устраняющие воспаление (Найз, Ибупрофен, Диклофенак);
- анальгетики (Кеторол);
- спазмолитики (Мидокалм).
Справочно. Показанием к операции является сильное сдавливание отделов мозга, явно выраженные неврологические расстройства, если положительный эффект от приема лекарственных препаратов не наблюдается.
Благодаря хирургическому вмешательству можно устранить чрезмерное давление на нервную ткань и привести в норму движение ликвора. Одной из проводимых операций является краниовертебральная декомпрессия, направленная на увеличение размеров задней черепной ямки.
Внимание. Декомпрессия относится к классу травматичных и рискованных операций. Согласно статистике, она приводит к осложнениям у каждого десятого пациента.
Риск летального исхода у прооперированных больных выше, чем у тех, кто операцию не переносил. Нейрохирурги стараются проводить такой тип вмешательства только в самых крайних случаях, когда налицо явные признаки сдавливания мозга. 
Иной вариант устранения последствий мальформации предполагает шунтирование, которое способно обеспечить отвод ликвора из черепной коробки. Благодаря имплантации специальных трубок жидкость перетекает в грудную и брюшную полости, ВЧД снижается.
В наиболее острых случаях требуется немедленная госпитализация больного и проведение всего спектра терапевтических, профилактических и коррекционных процедур.
Прогноз выживаемости
Продолжительность жизни зависит от типа аномалии и степени ее тяжести. Первый тип позволяет сделать благоприятный прогноз, поскольку симптоматика либо вовсе отсутствует, либо возникает после получения травм головы.
Если никаких проявлений болезни нет, то продолжительность жизни больных такая же, как и у здоровых людей.
Для пациентов со вторым типом аномалии прогноз хуже, она переносится тяжелее.
Иногда борьба с очаговой неврологической симптоматикой не приносит плодов даже при активном лечении медикаментами. В таком случае требуется хирургическое вмешательство, чтобы впоследствии изменения по неврологической части были менее выраженными.
Справочно. Третий и четвертый типы мальформации являются самыми тяжелыми для пациентов, прогноз зачастую неблагоприятен.
Заболевание затрагивает важные структуры мозга, у больного отмечаются пороки внутренних органов. Часто функции ствола мозга страдают настолько, что нарушения оказываются несовместимыми с жизнью.
Читайте также: