
Пояснично конечностная мышечная дистрофия
Конечностно-поясная мышечная дистрофия - это термин охватывает группу прогрессирующих наследственных миопатий, при которых поражаются главным образом мышцы тазового и плечевого пояса В конечном итоге также развивается атрофия и слабость мышц дистальных отделов конечностей. При некоторых формах формируется гипертрофия икроножных мышц и контрактура голеностопных суставов, в связи с чем может быть ошибочно диагностирована мышечная дистрофия Беккера.
Первые клинические проявления коненостно-поясной мышечной дистрофии редко возникают до среднего или старшего детского возраста либо могут быть отсрочены до раннего зрелого возраста. Боль в нижней части спины может служить первой жалобой и обусловлена формированием лордоза в связи со слабостью ягодичных мышц. Потеря способности к ходьбе (больные прикованы к инвалидному креслу) наблюдается обычно около 30 лет. Скорость прогрессирования заболевания варьирует в разных поколениях одной семьи, но одинакова у пациентов одного поколения.
Характерна слабость мышц-сгибателей и -разгибателей шеи, однако поражение мышц лица, языка и других мышц бульбарной группы встречается редко. По мере прогрессирования мышечной слабости и атрофии сухожильные рефлексы снижаются. Поражение сердца нехарактерно. Интеллект, как правило, не снижен. Дифференциальный диагноз конечностно-поясной мышечной дистрофии следует проводить с ювенильной спинальной мышечной атрофией (болезнь Кугельберга—Веландер), миастенией и метаболическими миопатиями:
В большинстве случаев конечностно-поясная мышечная дистрофия наследуется по аутосомно-рецесивному типу, в некоторых семьях наблюдается аутосомно-доминантный тип наследования. В последнем случае заболевание часто характеризуется доброкачественным течением и функциональные нарушения выражены незначительно.

ЭМГ и мышечная биопсия подтверждают диагноз мышечной дистрофии, но ни один из этих методов не специфичен настолько, чтобы установить точный диагноз без дополнительных клинических критериев. В некоторых случаях выявляется дефицит адхалена — дистрофинассоциированного гликопротеида сарколеммы; этот специфический дефект можно определить иммуноцитохимически на материале мышечной биопсии. Характерно повышение активности КФК в крови, однако она варьирует в разных семьях. ЭКГ, как правило, без изменений.
При одной аутосомно-доминантной форме конечностно-поясной мышечной дистрофии генетический дефект локализован на длинном плече хромосомы 5. При аутосомно-рецессивной форме ген картирован на длинном плече хромосомы 15. Продукт мутантного гена — дистрофинассоциированный протеин в саркогликановом комплексе (саркогликанопатия) ответствен за развитие некоторых случаев конечностно-поясной мышечной дистрофии с аутосомно-рецессивным типом наследования. Адхален — это а-саркогликан; встречаются другие формы конечностно-поясной мышечной дистрофии, обусловленные дефицитом b-, у- и q-саркогликана.
В нормальных (неизмененных) гладких мышцах а-саркогликан замещается на е-саркогликан.
Другая группа конечностно-поясных мышечных дистрофий обусловлена аллельными мутациями гена дисферлина (DYSF). Аномальный ген отвечает за синтез протеина, необходимого для структурной целости сарколеммы, хотя и не ассоциированного с комплексом дистрофин-гликопротеид. DYSF взаимодействует с кавеолином-3 или калпаином-3; дефицит DYSF может быть вторичным по отношению к дефектам этих продуктов других генов. Описаны формы с аутосомно-рецессивным (миопатия Миоши) и аутосомно-доминантным типом наследования.
Обе формы миопатий характеризуются медленно прогрессирующим течением с дебютом в подростковом или молодом возрасте и возможным поражением как дистальных, так и проксимальных мышц. Кардиомиопатия развивается редко. При дисферлинопатиях обнаружено повышение активности КФК в крови в несколько тысяч раз. Ультраструктурные изменения включают утолщение базальной пластинки над дефектами сарколеммы и замещение сарколеммы множественными слоями, состоящими из мелких пузырьков. Количество регенерирующих мышечных волокон превосходит таковое мышечных волокон с признаками дегенеративных изменений.
OMIM 253600
Наша команда профессионалов ответит на ваши вопросы
В 1954 году доктором Уалтоном и доктором Натрассом был впервые введен термин поясно-конечностые мышечные дистрофии (ПКМД илиLGMD), однако четкого понимания механизма заболевания не было. Это был универсальный термин, который использовали достаточно широко для разграничения людей с преобладающей конечностно-поясной слабостью от людей с другими типами дистрофий, таких, как Дюшена, Ландузи-Дежерина и др.
После первых проявлений заболевания больной может сохранить способность ходить более 20 лет. Мужчины и женщины болеют в равной мере. Некоторые врачи отмечают, что если болезнь появилась в детстве, то она прогрессирует быстрее и разрушительнее; если начало пришлось на юность или взрослый возраст, то обычно болезнь прогрессирует медленнее.
У некоторых больных в патологический процесс вовлекается сердце, но это бывает не так часто, как при других видах миодистрофий. Проблемы с сердцем могут проявиться в виде кардиомиопатий (слабость сердечной мышцы) или аритмий. В отдельных случаях заболевание само по себе может проявляться как кардиомиопатия.
По истечении тридцати или более лет к основным признакам болезни присоединяется дыхательная недостаточность. Боль не является характерной составляющей болезни. Но ограниченная подвижность иногда приводит к болезненности мышц и суставов. Интеллектуальные функции не страдают. Для большинства нозологических форм поясно-конечностных прогрессирующих мышечных дистрофий характерно повышение активности креатинфосфокиназы в плазме крови больных, которое может выявляться еще на доклинической стадии. Увеличение активности этого фермента может выявляться и у гетерозиготных носителей мутации в том или ином гене, которое, однако, не достигает такой степени, как у больных.
На ЭМГ и в мышечном биоптате выявляются признаки миопатии, мышцы истончены, часть волокон замещена жировой и соединительной тканью. В саркоплазме выявляются очаги фокального некроза. Ядра мышечных волокон центрально смещены, располагаются рядами или цепочками, вакуолизированы, с выраженным ядрышком. На поздних стадиях волокна теряют поперечную исчерченность, фрагментированы; иногда обнаруживаются только остатки миофибрилл.
При дифференциальной диагностике необходимо исключить воспалительные и метаболические миопатии, а также фенотипически сходные спинальные мышечные атрофии.
Частота всех поясно-конечностных мышечных дистрофий колеблется в различных популяциях от 5 до 70 больных на 1 миллион населения.В зависимости от типа наследования поясно-конечностные мышечные дистрофии разделяют на два типа.
К 1 типу относят нозологические формы с аутосомно-доминантным типом наследования. Для всех заболеваний этой группы известна локализация генов на хромосомах и для трех из них идентифицированы белковые продукты.
Ко второму типу относят формы заболевания, наследующиеся аутосомно- рецессивно. Первое описание ПКМД с аутосомно-рецессивным типом наследования проведено Kloepfer и Tally в 1958 году. Для большинства заболеваний этой группы идентифицированы гены, описаны основные типы их патологических мутаций и известен белковый продукт и его основные функции. На данный момент выделено 16 нозологических форм ПКМД с аутосомно-рецессивным наследованием и 7 с аутосомно-доминантным (Таблица 1), и их поиск продолжается.
Аутосомно-рецессивные формы ПКМД являются более распространенными, чем аутосомно- доминантные, которые составляют около 10% всех ПКМД. Различные группы населения часто имеют разные частоты различных типов ПКМД. Среди аутосомно-рецессивных форм наиболее распространены ПКМД 2A (от 30 % в Бразилии до 80% среди басков Испании от всех выявленных случаев) и ПКМД 2B (от 20% до 40% всех случаев). На группу саркогликанопатий (ПКМД 2C-2F) приходится еще около 20-25% всех случаев ПКМД, эта группа характеризуется тяжелым течением заболевания. Как и в случае с другими ПКМД, различные саркогликанопатии с различной частотой встречаются в разных популяциях. ПКМД 2C широко распространена в Тунисе; ПКМД 2D распространена в Европе, Соединенных Штатах и Бразилии, а ПКМД 2E и ПКМД 2F широко распространены в Бразилии. ПКМД 2I довольно распространена, особенно среди жителей Северной Европы. Недавние исследования показали, что больные с мутациями в этом гене составляют 6-38% случаев ПКМД. Остальные аутосомно-рецессивные формы ПКМД встречаются редко, и часто наблюдаются в изолированных группах населения.
Для LGMD 2I типа описаны частые мутации с.826C>A, с.229С>T, которые встречаются хотя бы на одной из хромосом более чем у 80% больных из РФ.
Для LGMD 2A типа описаны частые мутации с.550delA, с.598_612del15, которые встречаются хотя бы на одной из хромосом более чем у 83% больных из РФ.
По литературным данным для LGMD 2D типа описаны частые мутации, характерные для больных из РФ: c.229C>T и c.271G>A.
По литературным данным для LGMD 2L типа описаны частые мутации, характерные для европейцев: c.191dupA и c.2272C>T.
Таблица 1. Гены, ответственные за развитие различных форм ПКМД
Тип
заболе-
вания
OMIM
Тип
наследо-
вания
- Общее описание поясно-конечностной мышечной дистрофии (перевод материалов сайта mda.org)
- Организации и сообщества, посвящённые ПКМД. Диагностика ПКМД (клиники и врачи)
- Регистры пациентов с ПКМД
- День осведомлённости о ПКМД
- Статьи о ПКМД на нашем сайте
Что такое поясно-конечностная мышечная дистрофия?
Примечание переводчика. В разных источниках название варьируется: поясно-конечностная МД и конечностно-поясная МД.
Поясно-конечностная мышечная дистрофия (ПКМД) – это не одно общее заболевание. Это целая группа заболеваний, поражающих мышцы, в основном расположенные в районе бёдер и плеч. 
Плечевой пояс – это костная структура, которая окружает плечевую область.
(Информация из Википедии: плечевой пояс (пояс верхних конечностей) — совокупность костей (пары лопаток и ключиц) и мышц, обеспечивающих опору и движение верхних (передних) конечностей.)
Тазовый пояс – это костная структура, окружающая район бёдер.
По данным на конец 2012 года насчитывается более 20 различных подтипов ПКМД. Это сложная и постоянно развивающаяся область исследований.
Симптомы поясно-конечностной мышечной дистрофии
ПКМД, как и многие другие виды мышечных дистрофий, – это заболевание произвольно сокращающихся мышц. К таким мышцам относятся мышцы, используемые для движения конечностей, шеи, туловища и других частей тела, контролируемые волей человека. Через какое-то время мышечная слабость и атрофия могут привести к ограничению мобильности человека и неспособности поднять руки выше плеч.
При ПКМД непроизвольно сокращающиеся мышцы не повреждаются, за исключением сердца, которое является особым типом непроизвольно сокращающейся мышцы. Пищеварение, функции кишечника и мочевого пузыря, а также половые функции остаются в норме. ПКМД не влияет на мозг, интеллект и чувства. Пневмокардиальные осложнения могут возникнуть на поздних стадиях заболевания.
Причины возникновения поясно-конечностной мышечной дистрофии
ПКМД возникает из-за мутации в одном из (как минимум) 15 различных генов, из-за которой организм не может строить белки, необходимые для нормального функционирования мышц. Некоторые типы ПКМД – аутосомно-доминантные, что означает, что ПКМД была унаследована от одного из родителей. Другие типы ПКМД – аутосомно-рецессивные, возникающие в случае, если мутация в гене есть у обоих родителей.
Прогрессирование поясно-конечностной мышечной дистрофии
На настоящий момент прогрессирование каждого типа ПКМД не может быть предсказано точно, однако информация о генетической мутации, спровоцировавшей развитие ПКМД, может помочь. Некоторые формы ПКМД прогрессируют быстро (потеря способности ходить может произойти в течение небольшого количества лет) и влекут серьёзную степень обездвиженности. В то время как другие формы ПКМД прогрессируют медленно, причиняя минимальный вред двигательной способности.
ПКМД может стартовать в детстве, юношестве, раннем взрослом периоде или даже позже. Развитие ПКМД не зависит от пола человека.
Некоторые врачи приходят к выводу, что если ПКМД начинается в детстве, то прогрессирование обычно происходит быстрее, приводя к бОльшей степени инвалидности. Если же ПКМД начинается в юношестве или зрелом возрасте, то заболевание протекает в более слабой форме и прогрессирует медленнее.
Статус исследований поясно-конечностной мышечной дистрофии
Учёные, поддерживаемые Muscular Dystrophy Association, работают в нескольких различных направлениях, результаты которых могут иметь значение и для ПКМД. Направления исследований включают генную терапию, технологию пропуска экзона, технологию игнорирования стоп-кодона и блокирование миостатина.
Типы поясно-конечностной мышечной дистрофии
Ниже представлен список типов ПКМД.
Для типа 1 характерно наследование по доминантному признаку, т.е. требуется только одна мутация для проявления заболевания.
Для типа 2 характерно наследование по рецессивному признаку, т.е. требуется две мутации в гене – по одной от каждого родителя.
Некоторым типам ПКМД вместо чисел присвоены названия.
- миопатия Бетлема (мутация в гене collagen 6, доминантная)
- кальпаинопатия(мутация в гене calpain, рецессивная, другое название – LGMD2A)
- дисферлинопатия(мутация в гене dysferlin, рецессивная, другое название – LGMD2B)
- миофибриллярная миопатия (мутации в генах desmin, alpha-B crystallin, myotilin, ZASP, filamin C, BAG3 или SEPN1; все доминантные кроме desmin-типа, который может быть как доминантным, так и рецессивным)
- саркогликанопатии (мутация в гене sarcoglycan; рецессивная; другие названия – LGMD2C, LGMD2D, LGMD2E, LGMD2F)
- ZASP-связанные миопатии (мутация в гене ZASP; доминантная; является формой миофибриллярной миопатии)
- LGMD1A / ПКМД1А (мутация в гене myotilin)
- LGMD1B / ПКМД1B (мутация в гене lamin A/C)
- LGMD1C / ПКМД1С (мутация в гене caveolin)
- LGMD1D / ПКМД1D (мутация в гене DNAJB6)
- LGMD1E / ПКМД1E, также называемая десминовая миопатия – тип миофибриллярной миопатии (мутация в гене desmin)
- LGMD1F / ПКМД1F (мутация на 7-й хромосоме)
- LGMD1G / ПКМД1G (мутация на 4-й хромосоме)
- LGMD1H / ПКМД1H (мутация на 3-й хромосоме)
- LGMD2A / ПКМД2A(мутация в гене calpain)
- LGMD2B / ПКМД2B (мутация в гене dysferlin)
- LGMD2C / ПКМД2C, также называется SCARMD1 (мутация в гене gamma sarcoglycan)
- LGMD2D / ПКМД2D, также называется SCARMD2 (мутация в гене alpha sarcoglycan)
- LGMD2E / ПКМД2E (мутация в гене beta sarcoglycan)
- LGMD2F / ПКМД2F (мутация в гене delta sarcoglycan)
- LGMD2G / ПКМД2G (мутация в гене telethonin)
- LGMD2H / ПКМД2H (мутация в гене TRIM32)
- LGMD2I / ПКМД2I (мутация в гене FKRP)
- LGMD2J / ПКМД2J (мутация в гене titin)
- LGMD2K / ПКМД2K (мутация в гене POMT1)
- LGMD2L / ПКМД2L (мутация в гене ANO5)
- LGMD2M / ПКМД2M (мутация в гене fukutin)
- LGMD2N / ПКМД2N (мутация в гене POMT2)
- LGMD2O / ПКМД2O (мутация в гене POMGnT1)
- LGMD2Q / ПКМД2Q (мутация в гене plectin)
Организации и сообщества, посвящённые ПКМД. Диагностика ПКМД
- Общий список клиник и врачей, занимающихся нервно-мышечными заболеваниями, – по ссылке.
- Список зарубежных организаций по ПКМД.
- Список зарубежных сообществ по ПКМД.
- Закрытая Facebook-группа CureLGMD2I
- Информация о клиническом испытании PF-06252616 для LGMD2I
- Фонд The CureLGMD2i
- Список исследований по мутациям в гене FKPR
- Исследовательский фонд LGMD2I Research Fund
Объявление от администраторов сайта
Приглашаем желающих курировать раздел о поясно-конечностной мышечной дистрофии.
Важно! Регистры (реестры) пациентов с ПКМД
- Международный регистр пациентов с ПКМД 2А (LGMD2A, кальпаинопатия)
- Международные регистры пациентов с неопределённой формой МД, с формой ПКМД2B (LGMD2B, дисферлинопатия, миопатия Миоши), а также с другими формами миопатий (ПКМД/LGMD/Эрба-Рота, HIBM, Помпе, Бетлема, EDMD, ЛЛПМД/Ландузи-Дежерина, Дюшенна/Беккера)
- Международный регистр пациентов с ПКМД 2D (LGMD2D, ПКМД 2Д, мутация в гене альфа-саркогликан)
- Международный регистр пациентов с ПКМД 2I (LGMD2I, мутация в гене FKRP – fukutin-related protein)
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку ПКМД является редким (орфанным) заболеванием.
Для этого в разных странах ведутся регистры (реестры) пациентов с ПКМД — базы данных по генетической и клинической информации о людях, страдающих ПКМД и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных данным заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
День осведомлённости о ПКМД

30 сентября – день осведомлённости о поясно-конечностной мышечной дистрофии.
Страница о дне осведомлённости на англоязычном информационном портале сообщества ПКМД
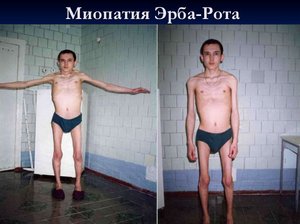
В отечественной литературе эта форма известна как ювенильная миопатия Эрба, однако в иностранной литературе она описана как конечностно-поясная миодистрофия. Встречается с частотой 1,5:100 000.
Ряд авторов выделяют аутосомно-рецессивную форму миодиетрофии с ранним началом и тяжелым течением, напоминающую миодистрофию Дюшенна (так называемая псевдодюшенновская форма). В отличие от Х-сцепленной формы Дюшенна при данной форме ЭКГ имеет нормальный вид, хотя патология сердца и может развиться в поздней стадии заболевания. Частота этой формы составляет около 1/4 от частоты истинной формы Дюшенна.
Начало заболевания чаще всего относится к середине второго десятилетия жизни (14 — 16 лет), что и обусловливает одно из названий — ювенильная форма, однако первые симптомы могут проявиться до 10 лет, а иногда и после 30 (так называемая поздняя миопатия).
Для миодиетрофии Эрба характерны умеренные псевдогипертрофии, сухожильные ретракции, контрактуры. Могут иметь место концевые атрофии. Интеллекту больных не страдает, сердечная мышца большей частью также не поражена. Уровень ферментов в сыворотке крови обычно повышается, особенно на ранних стадиях процесса, однако далеко не столь резко, как при Х-сцепленных формах миодиетрофии.
Электромиография выявляет мышечный тип поражения со снижением амплитуды биопотенциалов и сохранной частотой, однако, по наблюдениям некоторых авторов [McComas A. et al., 1971], в дистальных мышцах могут наблюдаться признаки денервации.
Дистрофия Эрба — наследственное заболевание с аутосомно-рецессивным типом передачи, оба пола страдают одинаково часто без особой разницы в тяжести проявлений.
Приводим характерную для этого страдания родословную семьи С-ких.
Обращает внимание, что мать и отец родом из одной деревни — это предполагает кровное родство. В данной семье все трое детей (две девочки и один мальчик) являются гомозиготными носителями мутантного гена, хотя по законам Менделя должны заболеть 25% детей.
Родословная семьи С-ких

Миодистрофия Эрба. Обозначения те же, что и на рисунке
Родословная семьи Д-вых.
В следующем наблюдении можно проследить внутрисемейный клинический полиморфизм, обусловленный усилением экспрессивности экзогенными факторами.
Больной Д., 30 лет, находился в клинике нервных болезней. Жалобы при поступлении на снижение силы в руках и ногах, нарушение походки, затруднение ходьбы по лестнице. В 16-летнем возрасте впервые появилась слабость в ногах, затем вскоре в руках, стало трудно поднимать руки, изменилась походка.
Соматический статус пробанда без особенностей. В неврологическом статусе — легкое расходящееся косоглазие (в анамнезе в школьные годы черепно-мозговая травма), легкая слабость круговой мышцы глаза с обеих сторон. Ограничен объем активных движений в проксимальных отделах рук и ног со снижением мышечной силы до 3 баллов.
ЭКГ — без патологии.
Общие анализы крови и мочи без отклонений от нормы.
Активность КФК — 1,2 ЕД, Ф-1,6-Ф-альдолазы — 12 ЕД, ЛДГ — 225 ЕД. Белок, альбумин, неорганический фосфор, билирубин, мочевица, глюкоза в сыворотке крови — без отклонений от нормы.
Начало заболевания с 16-летнего возраста, относительно благоприятное течение, проксимальный вялый тетрапарез с характерным изменением походки, крыловидными лопатками и исчезновением сухожильных рефлексов, а также характер ЭМГ, незначительная гиперферментемия — позволяют поставить диагноз миодиетрофии Эрба.
В семье имеется еще один больной со значительно более мягкой формой заболевания, что демонстрирует внутрисемейное различие в результате разной экспрессивности. Обращает внимание, что у пробанда с более тяжелой формой заболевания в анамнезе — черепно-мозговая травма.
Необходимо иметь в виду также эндокринные формы миопатического синдрома, лекарственные (стероидные, делагил), токсические (алкоголь и др.) и карциноматозные миопатии при поражении мышц в пожилом возрасте.
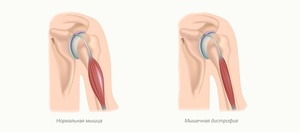
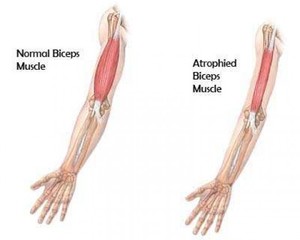
Мышечные дистрофии представляют собой группу наследственных первичных заболеваний мышц, характеризующихся дегенерацией мышечных волокон и мышечной слабостью.
Мышечная слабость при мышечной дистрофии
Классификация этих состояний традиционно основывалась на:
- патологических,
- клинических,
- наследственных паттернах.
Даже при том, что есть некоторые общие признаки, относящиеся к мышечной слабости, определенные синдромы показывают некоторые характерные клинические проявления.
Мышечная слабость, возникающая при всех различных типах мышечной дистрофии, имеет определенную характеристику. Прежде всего, пациент ощущает слабость с обеих сторон тела. Как правило, при мышечной слабости отсутствуют болевые ощущения, но мышцы чувствительны к прикосновениям. Известно, что при некоторых типах дистрофий, затрагивающих основные мышцы конечностей, возникают скованность и судороги (хотя тяжелые формы судорог довольно необычны).
Течение и степень тяжести заболевания, а также его прогноз зависят от конкретного типа дистрофии, от которой страдает человек, следовательно, необходим точный диагноз. Тщательное изучение истории болезни и соответствующие лабораторные анализы имеют решающее значение для установления диагноза и планирования терапевтического процесса.
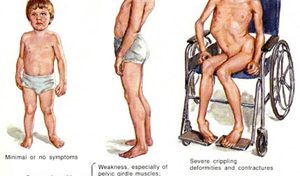
Симптомы мышечной дистрофии Дюшенна обычно появляются в возрасте до 6 лет. Слабость начинается в области верхних конечностей и таза. Дети, страдающие данным заболеванием, часто падают, имеют проблемы с двигательными навыками (бег, прыжки) и проявляют псевдогипертрофию. Симптомы могут также включать усталость, трудности в обучении (IQ может быть ниже 75) и возможную умственную отсталость.

Мышечная дистрофия Беккера менее серьезна, чем мышечная дистрофия Дюшенна, поэтому основное различие заключается в том, что люди с этим заболеванием могут ходить в 16 лет (а некоторые продолжают делать это и в пожилом возрасте).
У них могут быть:
- судороги в мышцах,
- определенные трудности с координацией.
Наиболее выраженным симптомом миотонической мышечной дистрофии является неспособность расслабить мышцы после внезапного сокращения. Это состояние поражает как женщин, так и мужчин в возрасте от 20 до 30 лет.
Другие клинические проявления включают:
- проблемы со зрением,
- трудности с глотанием,
- сильное снижение веса,
- облысение,
- сердечные заболевания,
- атрофию яичек.
Термин "врожденная мышечная дистрофия" используется для группы мышечных дистрофий, объединенных тем фактом, что мышечная слабость начинается в младенчестве или в раннем детстве (обычно в возрасте до двух лет). Из-за этих состояний ребенок может казаться "дискетным", и позже дети медленно достигают таких основных двигательных показателей, как сидение, переворачивание или ходьба. Некоторые из более редких форм врожденной мышечной дистрофии также сопровождаются умственной отсталостью или неспособностью к обучению.
Ранние признаки мышечной дистрофии Эмери-Дрейфуса включают ходьбу на пальцах ног из-за жесткости ахиллова сухожилия в пятках, трудности сгибания в локтях и возможность обморока из-за аномалий сердца.
Мышечная дистрофия конечностей, как и другие мышечные дистрофии, в первую очередь представляет собой расстройство произвольных мышц. Люди с этим заболеванием сталкиваются со слабостью мышц бедер и ног.
Встройте "Правду.Ру" в свой информационный поток, если хотите получать оперативные комментарии и новости:
Подпишитесь на наш канал в Яндекс.Дзен или в Яндекс.Чат
Добавьте "Правду.Ру" в свои источники в Яндекс.Новости или News.Google
Также будем рады вам в наших сообществах во ВКонтакте, Фейсбуке, Твиттере, Одноклассниках.

Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Распространенность заболевания
Миопатия относится к редким и неизлечимым заболеваниям. Трудно вывести точную статистику, поскольку она объединяет различные болезни. Согласно некоторым исследованиям, около 1 из 3 500 человек страдают от этого заболевания.
Например:
![]()
Миопатия Дюшенна затрагивает приблизительно одного ребенка (мальчика) из 3500.- Миопатия Беккера касается 1 мальчика из 18 000.
- Фазио-скапулогумаральная дистрофия поражает около 1 из 20 000 взрослых людей.
- Болезнь Эмери-Дрейфус затрагивает 1 из 300 000 человек, вызывает ретракцию сухожилия и нарушение сердечной мышцы
Частота и тип заболеваний зависит от конкретной страны:
Причины заболевания и лечение
Причиной данной патологии являются генетические заболевания, то есть дефект (или мутация) гена, необходимого для нормального развития мышц. Когда этот ген мутирует, мышцы больше не в состоянии нормально функционировать — они теряют свой силовой потенциал и в результате атрофируются.
Например:
- Миопатия Дюшенна связана с дефицитом дистрофина — белка, расположенного под мембраной мышечных клеток, который играет роль в сокращении мышц.
- Почти у половины врожденных МД причиной является дефицит мерозина — белка, составляющего мембрану клеток мышцы.
Как правило, МД передается рецессивно. Другими словами, для того чтобы болезнь выражалась, оба родителя должны быть носителями и передавать ребенку ненормальный ген. Болезнь не проявляется у родителей по той причине, что у каждого из них есть только один аномальный ген, а не два. Для нормального функционирования мышц достаточно одного нормального гена.
Кроме того, некоторые формы миопатии затрагивают только мальчиков: это миопатия Дюшенна и Беккера. В обоих случаях ген, участвующий в этих двух заболеваниях, расположен в Х-хромосоме, которая существует в единственной копии у мужского пола.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
Исследования и прогресс
С 2005 года для лечения пациентов с развивающимся поражением мышц все чаще используются стволовые клетки. Для лечения мышечной дистрофии этим методом могут быть рассмотрены различные варианты заболевания, такие как: мышечные дистрофии Дюшенна, Беккера, миопатия поясничного и плечевого отдела.
Целью лечения является регенерация потерянных и поврежденных мышечных волокон с использованием регенеративного потенциала стволовых клеток. Для этого большое количество стволовых клеток вводится при помощи нескольких внутривенных и внутримышечных инъекций, что позволяет лучше нацеливать терапию именно на пораженную группу мышц.
Возможный прогресс
Терапия с применением стволовых клеток может обеспечить улучшение в плане мышечной массы, силы, движений, баланса, тремора и ригидности мышц. Стволовые клетки также могут замедлить будущую потерю мышечного объема и уменьшить симптомы.
Важно отметить, что лечение не является окончательным лекарством от этого заболевания и никоим образом не может решить проблему потери мышечных волокон. По этой причине прогресс после такого лечения не может быть постоянным. Исследования в этой области все еще ведутся.
Семейства заболевания
Обычно существуют два основных семейства МД:
- Мышечна врожденная дистрофия (ВМД), которая выражается в первые 6 месяцев жизни, сопровождает около десяти форм патологий переменной тяжести, включая ВМД с первичной недостаточностью мерозина, синдром Ульриха и Уокера-Варбурга;
- Мышечные дистрофии, появляющиеся в детстве или в зрелом возрасте:
![]()
Миопатия Дюшенна- Миопатия Беккера
- Миопатия Эмери-Дрейфуса (существует несколько форм)
- миопатия Ландузи-Дежерина
- Так называемая миопатия поясничного отдела — затрагивает мышцы вокруг плеч и бедер.
- Миотонические дистрофии (типы I и II), которые включают болезнь Штейнтера. Они характеризуются миотонией — когда мышцы не могут нормально расслабиться после сокращения.
- Окулофарингеальная миопатия
Эволюция дистрофии
Эволюция (развитие заболевания) МД сильно варьируется от одной формы к другой, а также от одного человека к другому. Некоторые формы быстро развиваются, что приводит к ранней утрате подвижности и ходьбе, а иногда и к смертельным сердечным или респираторным осложнениям, в то время как другие развиваются очень медленно — в течение десятилетий. Большинство врожденных мышечных дистрофий, например, которые мало выражены или почти незаметны, позже могут проявятся внезапно и с серьезными последствиями.
Возможные осложнения
Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Таким образом, сердечные осложнения довольно распространены, особенно у мальчиков с мышечной дистрофией Дюшенна.
Кроме того, дегенерация мышц заставляет тело и суставы деформироваться постепенно: на фоне этого у больных может развиваться сколиоз. Часто наблюдается сокращение мышц и сухожилий, что приводит к их стягиванию. Все эти нарушения приводят к деформации суставов: ноги и руки повернуты внутрь и вниз, деформируются колени или локти.
Также известно, что болезнь сопровождается тревожными или депрессивными расстройствами, поэтому больным требуется много внимания и поддержки, в первую очередь со стороны близких.
Читайте также: