
Спинальная мышечная атрофия беременность
Спинальная мышечная атрофия или СМА – генетически обусловленная патология, обнаруживаемая у младенцев, детей дошкольного возраста, подростков и взрослых и сопровождающаяся равносторонней атрофией нейронов спинномозговых передних рогов и корешков периферических нервов, что приводит к снижению мышечного тонуса и прогрессирующему параличу. В первом случае медики вынуждены констатировать тот факт, что ребенок никогда не сможет самостоятельно стоять, сидеть и ходить. В остальных он будет постепенно утрачивать эти способности и однажды окажется прикованным к инвалидному креслу.


Что такое спинальная мышечная атрофия и ее виды
Под этим термином объединяется несколько различных видов наследственных заболеваний, сопровождающихся ограничением двигательных способностей. Этим и объясняется тот факт, что в части случаев нарушения обнаруживаются не в младенческом возрасте, а у подростков или уже зрелых людей.
Впервые заболевание было описано в 1891 г. Г. Верднигом и в 1892 г. было выделено в отдельную нозологическую единицу Дж. Хоффманом, благодаря стараниям которых и получила свое второе название. Примерно через полвека Е. Кугелбергом и Л. Веландером была открыта другая подобная болезнь, развивающаяся в более позднем возрасте и отличающаяся более благоприятным течением.
Различают следующие формы патологии:
- СМА 0;
- СМА 1 (тяжелая форма);
- СМА 2 (промежуточная форма);
- СМА 3 (легкая форма);
- СМА 4 (поздняя форма).
Все их объединяет то, что причина их возникновения кроется в мутации рецессивного гена 5 хромосомы SMN. Это приводит к сбоям в продукции протеинов в организме, являющихся строительным материалом всех клеток. В результате страдают мотонейроны спинного мозга и постепенно разрушаются. Поскольку без них невозможна передача нервных импульсов к мышечным волокнам, они постепенно атрофируются, что становится причиной утраты способности двигаться.

К счастью, даже при наличии у обоих родителей мутации гена SMNу них с 75% вероятностью может родиться здоровый ребенок. Но практически всегда он также будет носителем этого гена. Поэтому при планировании беременности стоит проходить генетическое исследование, особенно при наличии случаев СМА в семье.
Это врожденная болезнь, признаки которой обнаруживаются обычно еще в роддоме. Она встречается редко и ее часто объединяются со СМА-1. Для этого вида типично абсолютное отсутствие подвижности, слабость мышц, отсутствие сухожильных рефлексов и ограничение функциональности коленных суставов. С первых дней жизни ребенок страдает от нарушения дыхания.
Спинально-мышечную атрофию важно дифференцировать с перинатальной энцефалопатией и родовыми травмами, но если при них состояние детей постепенно улучшается, то при СМА оно не меняется. Более того часто присоединяются осложнения, которые практически всегда приводят к смерти младенцев в течение первого месяца жизни.

Этот тип течения спинальной мышечной атрофии характеризуется очень тяжелым протеканием. Обычно она обнаруживается до 6-ти месяцев и сопровождается слабостью мышц, периодическими спазмами, что сложно заметить в связи с особенностями анатомии детей первого года жизни (присутствия ярко-выраженной подкожно-жировой клетчатки).
Также заболевание проявляется регулярно пробегающей по языку дрожью, снижением рвотного, сосательного, глотательного рефлексов. Это приводит к возникновению серьезных трудностей при кормлении. Присутствует нарушение слюноотделения, кашель. Ребенок часто громко кричит.
Эта форма спинально-мышечной атрофии может сопровождаться олигофренией и врожденными пороками сердца. Дети подвержены тяжелым нарушениям дыхания, развитию воспаления легких. В связи с этим более половины детей не доживает до 2 лет и только 10% могут отметить свой 5-летний юбилей. Причиной смерти становятся пневмония, остановка сердца или дыхательная недостаточность.
Заболевание обнаруживается у детей от 6 месяцев до 1,5–2 лет. Поэтому такую форму СМА часто называют поздней младенческой. Для нее типично:
- слабость и дрожь в мышцах;
- тремор пальцев, языка;
- скованность движений, обусловленная ограничением подвижности конечностей;
- задержка развития;
- недобор веса.
Дети с таким диагнозом способны самостоятельно сидеть, играть, есть, но стоять и передвигаться нет. К сожалению, патология склонна прогрессировать, что приводит к постепенному ослаблению мышц груди и шеи, следствием чего становится невозможность удерживать голову прямо и часто она безвольно свисает. Затем пропадают сухожильные рефлексы, слабеет голос и отмечаются нарушения акта глотания.
Длительность жизни при таком диагнозе составляет около 10–12 лет. Но треть больных погибает в возрасте до 4-х лет.
Спинальную мышечную атрофию этого вида диагностируют обычно после 2 лет. Она так же проявляется слабостью мышц, но не в такой степени как при СМА 1 или даже СМА 2. Больные могут самостоятельно стоять, но только в течение короткого периода времени. В связи с атрофией мышц это дается им с трудом.
Несмотря на имеющееся заболевание, до 10–12 лет ребенок развивается нормально, что может ввести его родных в заблуждение и вызвать сомнения в правильности поставленного диагноза. Но, достигая этого временного рубежа, возникают первые признаки СМА. Ребенок начинает спотыкаться чаще обычного, падает и не может выполнять физическую работу или заниматься спортом, часто сталкивается с переломами. Постепенно бег, а затем и ходьба даются все сложнее из-за возникновения ограничения подвижности суставов. Впоследствии подросток теряет способность передвигаться без инвалидного кресла.
Прогрессирование патологии приводит к возникновению тяжелого сколиоза, что влечет за собой изменение формы грудной клетки и появление трудностей при дыхании. Именно в этом таится главная угроза болезни для жизни.
К этому типу заболевания относят несколько разных не влияющих на продолжительность жизни, но приводящих к инвалидизации амиотрофий:
- бульбоспинальную Кеннеди;
- дистальную Дюшена-Арана;
- перонеальную Вюльпиана.
Их объединяет то, что первые клинические признаки заболевания проявляются в период от 16 до 60 лет, чаще в 35–40 лет. Это сопровождается угасанием сухожильных рефлексов и заметными спазмами мышц. При атрофии Дюшена-Арана сильнее всего страдают кисти, а для болезни Вюльпиана характерно изменение формы лопаток на крыловидную.
Симптомы СМА
Различные разновидности заболевания объединяет череда общих проявлений, хотя каждая из них имеет и специфичные симптомы. К числу общих признаков принадлежат:
- нарастающая мышечная слабость и постепенная атрофия;
- при тех видах спинальной мышечной атрофии, что обнаруживаются после года или двух лет, наблюдается деградация имеющихся физических достижений, к примеру, способности бегать, ходить;
- тремор пальцев, языка;
- искривление позвоночника;
- частое сохранение нормального психического развития и умственных способностей.
Статистика показывает, что чаще СМА поражает мальчиков.
Диагностика
Предельно информативным методом диагностики СМА считается генетический анализ. Его можно проводить ребенку и взрослому в любом возрасте, а с целью ранней диагностики его выполнение возможно еще на этапе внутриутробного развития. При невозможности проведения анализа ДНК и для окончательного подтверждения диагноза назначаются:
- биохимический анализ крови;
- гистологический анализ мышечных волокон;
- МРТ;
- электромиография;
- микроскопия спинного мозга;
- тандемная масс-спектрометрия.

Лечение спинально-мышечная атрофия
Больным назначается комплексная консервативная терапия, направленная на улучшение способности нервных импульсов проходить к мышцам и работы головного мозга. В этих целях рекомендуется прием:
- ноотропов;
- препаратов α-липоевой кислоты, ацетил-L-карнитина, α-глицерофосфохолина;
- витаминных комплексов, включающих, прежде всего, витамины группы В;
- средств, улучшающих обмен веществ.

Сегодня в разработке находятся специфические лекарственные средства, способные воздействовать на причину развития СМА – дефицит ряда белков. Но в данный момент они находятся на стадии испытаний. Пока что единственным способом хотя бы частично обеспечить организм необходимыми белками является соблюдение специальной диеты. Она подразумевает употребление продуктов, богатых на аминокислоты, а именно зерновых культур, орехов, кисломолочных продуктов, рыбы, мяса. Весьма полезно включение в меню шпината, брокколи, грейпфрутов. Особенно ценны блюда из бурого риса и овса.
Для поддержания мышечного тонуса рекомендованы:
- занятия ЛФК;
- массаж;
- физиотерапевтическое лечение;
- нейромышечная стимуляция.
Современная медицина способна помочь пациентам с СМА за счет выравнивания позвоночника. Вы можете существенно повысить качество жизни и избавиться от болей с помощью хирургического лечения нейромышечного сколиоза. Наши спинальные хирурги способны грамотно провести операцию с учетом всех особенностей пациента и добиться предельно высоких результатов. Цены наших услуг приведены в прайсе.
Суть хирургического лечения нейромышечного сколиоза заключается в выполнении многоуровневой фиксации позвоночника с помощью специальных конструкций. Это предполагает изменение и закрепление в максимально приближенном к нормальному положению каждого сегмента искривленной части позвоночного столба.
Многоуровневая фиксация реализуется за счет установки многочисленных опорных элементов и выбора в качестве опорных точек крестца и таза и позвонков верхнегрудного отдела. Но часто ее проведение требуется практически по всей длине позвоночника, так как у больных спинальной мышечной атрофией сколиотические деформации достигают предельно тяжелых форм.
Она позволяет не только практически полностью выровнять позвоночник, но и равномерно распределить нагрузку на него, а также надежно удерживать его в новом положении. Благодаря этому больной избавляется от выраженного комфорта во время сидения и лежания, решаются психологические проблемы, спровоцированные выраженной деформацией позвоночного столба. Но главное достоинство операции заключается в устранении негативного влияния сколиоза на легкие и другие внутренние органы.
Стоимость коррекции сколиоза при СМА от 640 000 руб и зависит от:
— Тяжести заболевания (сколько времени пациент проведен в стационаре после операции)
— Фирмы производителя имплантов;
— Клиники (где будет проведена операция) и класса палаты.
Цена включает в себя:
— Прибывание в клинике до и после операции;
— Импланты.
— Операцию;
— Наркоз;
— Нейрофизиологический мониторинг
— Наблюдение и консультация на период реабилитации.
Все услуги клиники и стоимость приведены в прайсе.
На первичной консультации ответим на все интересующие вас вопросы, точно определим возможные риски и потенциальную пользу хирургического лечения и подарим вашему ребенку если не возможность ходить, то уверенно сидеть без болей и психологического дискомфорта.



Дефицит важного белка, который не вырабатывает ген SMN1, приводит к атрофии (потере жизнеспособности и уменьшению размера) мышц, и в результате человек постепенно теряет способность ходить, удерживать и управлять собственным телом, самостоятельно сидеть, есть, глотать и дышать. Упрощенный механизм СМА можно увидеть на схеме.

К счастью, у человека есть еще ген SMN2, который дублирует SMN1. Он производит нефункциональный белок и небольшое количество нормального белка, который позволяет мотонейронам человека выживать на очень голодном пайке. В результате человек со СМА сохраняет какую то часть своих двигательных, глотательных и дыхательных функций.
От чего зависит тип СМА и тяжесть болезни?
Если максимально упростить картину, то у человека может быть разное число копий гена SMN2. Теоретически, чем больше у пациента со СМА таких копий, которые вырабатывают маленькое количество нужного мотонейронам белка, тем легче будет протекать заболевание, человек сохранит больше жизненно важных функций и дольше проживет. На практике есть слишком много нюансов, чтобы напрямую связывать тяжесть болезней с числом копий SMN2.
При этом у людей признаки СМА появляются в разное время: от первых дней жизни до взрослого возраста.
Существует четыре типа спинальной мышечной атрофии. Чем раньше начинается заболевание, тем тяжелее оно протекает. В 95 % случаев причиной СМА становится делеция (выпадение) участка генетического кода.
По международным стандартам типы СМА различаются несколькими важнейшими признаками:

Болезнь Верднига-Гофманна (СМА 1), возраст дебюта заболевания от 0 до 6 месяцев, максимальная (двигательная) функция у таких детей: сами не могут сидеть. При естественном течении заболевания 92 % этих деток погибает до возраста двух лет.

Болезнь Дубовица (СМА 2). Дебют заболевания происходит с 6 до 18 месяцев. Такие пациенты никогда не могут самостоятельно стоять, они могут самостоятельно сидеть. Продолжительность их жизни может быть больше двух лет

Болезнь Кугельберга-Велендер (СМА 3). Дебют заболевания происходит позже 18 месяцев, такие пациенты развиваются и нормально ходят, но в какой-то момент заболевание начинает прогрессировать. Они перестают самостоятельно передвигаться и садятся. Хотя продолжительность жизни у них тоже сильно ограничена, но они могут прожить несколько десятилетий.

Четвертый тип мы почти не видим в детской клинике. Такие пациенты дебютируют в подростковом и взрослом возрасте, они могут потерять способность к самостоятельному передвижению. Продолжительность жизни — десятилетия.
Возраст дебюта заболевания
Средняя продолжитель-ность жизни
Тип 1
Задержка моторного развития и набора веса, слабый кашель, тремор рук, контрактуры, сколиоз и деформация грудной клетки.
Тип 3
Старше 18 месяцев
Мышечная слабость различной степени выраженности, крампи (судороги в икроножных мышцах), контрактуры и гипермобильность суставов, потеря способности ходить по мере прогрессирования заболевания
Тип 4
В подростковом или взрослом возрасте
Могут потерять способность к самостоятельному передвижению
Прогрессирующая проксимальная мышечная слабость, снижение сухожильных рефлексов, фасцикуляции
Чуть более подробно и наглядно об особенностях разных типов СМА можно посмотреть по ссылке
Почему рождается ребенок со СМА?
Спинальная мышечная атрофия — это генетическое заболевание. Каждый сороковой житель планеты — здоровый носитель СМА. Чтобы в семье родился ребенок со СМА, нужно, чтобы такими носителями были оба родителя. В этом случае в 25% случаях ребенок родится здоровым, в 50% случаях он получает мутацию гена SMN1 от папы или от мамы и тоже становится просто носителем, а еще в 25% случаях он наследует мутацию гена SMN1 от обоих родителей и рождается с этой болезнью.
Отметим, что у здоровых носителей мутации гена SMN1 есть один полноценный ген и один ген с мутацией. Наличие одного полноценного гена достаточно для выработки необходимого количества нормального белка и правильного функционирования мотонейронов. Так что носитель от человека без поломки этого гена отличается лишь потенциальным риском рождения ребенка со СМА.
Более наглядно это показано на схеме
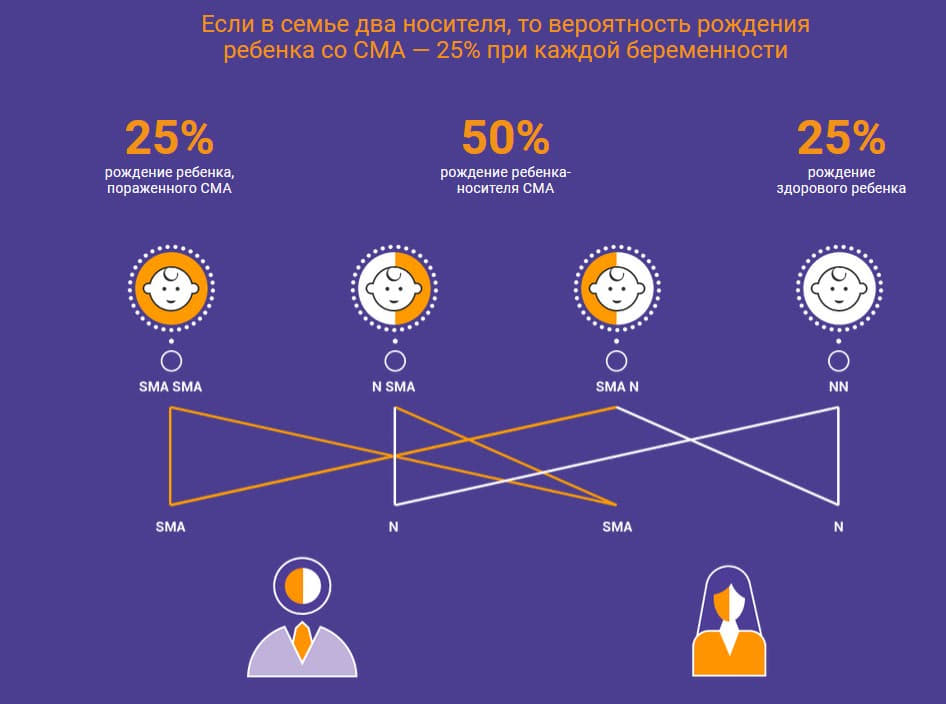
К сожалению, в реальности генетические особенности человека срабатывают не по математическому принципу.
Кто и как ставит диагноз "Спинальная мышечная атрофия"
Поставить клинический диагноз СМА врач может после неврологического осмотра пациента с проверкой рефлексов и мышечного тонуса. Еще нужно провести внешний осмотр пациента и сделать биохимический тест.
После этого человек должен пройти еще несколько тестов. Генетическое исследование должно подтвердить или опровергнуть первоначальный клинический диагноз.
Тесты также нужны для уточнения прогноза заболевания и уточнения носительства мутаций гена SMN1 у родителей ребенка.
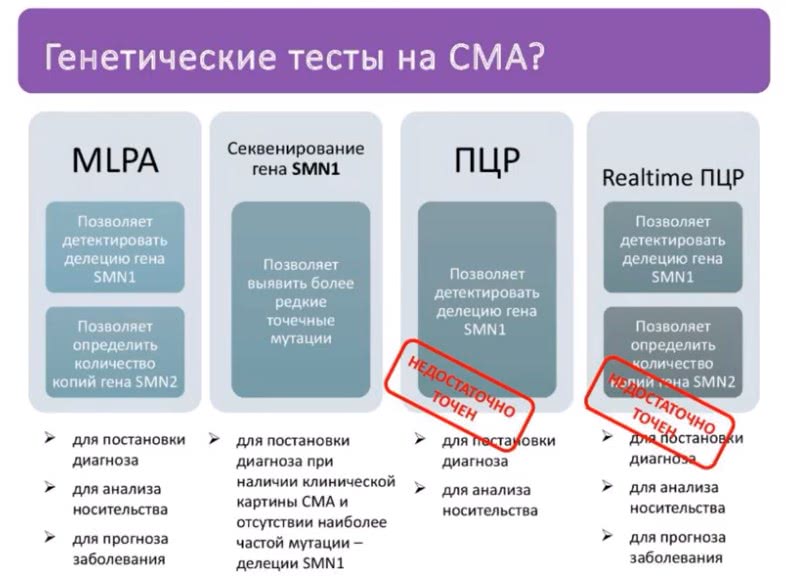
Подробнее о назначении каждого теста можно узнать, посмотрев на изображение.
Лучший тест на сегодня — MLPA. Он подтверждает причину СМА в 95 % случаев. Для оставшихся 5 % нужно тест секвенирование гена SMN1 (он нужен, если первый тест не подтвердил диагноз, а у пациента есть клинические проявления СМА).
СМА и беременность
Если в семье уже есть ребенок со СМА или один из будущих родителей — носитель или пациент со СМА, необходимо заранее провести несколько тестов .
Если два основных генетических исследования (MPLA и секвенирование) показывают, что второй родитель тоже носитель мутации гена SMN1, нужно решить, каким образом запланировать и провести беременность, чтобы минимизировать риск появления ребенка со СМА.
Если супружеская пара выбирает естественный путь для зачатия, то она должна пройти пренатальную инвазивную диагностику после десятой недели беременности. Для этого прокалывают живот и делают забор материала у плода. В современных условиях процедура достаточно безопасна и для будущей мамы, и для плода. Существует минимальный риск выкидыша, но он гораздо ниже, чем вероятность рождения ребенка со СМА.
Если супруги предпочитают ЭКО, то они могут пойти двумя путями:
или из нескольких зародышей отобрать здорового, или воспользоваться услугами донора — неносителя СМА. В этом случае донора нужно дополнительно исследовать на наличие мутации в гене SMN1.
При отборе эмбрионов риск возможного повреждения каждого эмбриона стремится к нулю. Отобранный здоровый эмбрион замораживают и подсаживают в матку.
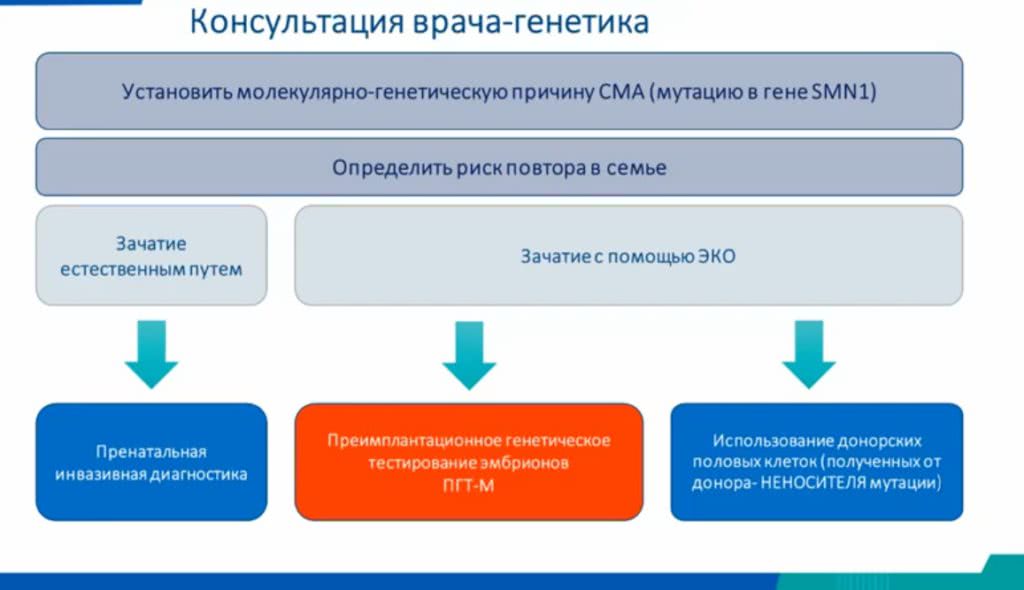
Более подробно о беременности при СМА можно узнать, посмотрев запись вебинара Натальи Ветровой.
Патогенетическая терапия при СМА
В настоящее время в мире есть два уже одобренных препарата для лечения СМА — Нусинерсен (Спинраза) и AVXS -101 (Золгенсма). Ожидается, что в ближайшее время на рынке появится третий препарат RG7916 (Рисдиплам).
Нусинерсен предназначен для пожизненного приема, в случае с Золгенсма речь идет об однократной дозе, но оба этих препарата работают неодинаково. Один решает проблему мышечной слабости, другой — препарат, который модифицирует генные клетки в целом.
Главная задача этих препаратов — восполнить дефицит белка из-за не работающего гена SMN1. О проведенных клинических испытаниях препаратов и о последних новостях лекарственной терапии при СМА можно прочитать в разделе Клинические исследования и лекарства .
Краткий обзор о принципах работы патогенетической терапии при СМА можно прочитать по ссылке .
-
13 декабря 2019
Публичная оферта о заключении договора пожертвования
1. ПРЕДМЕТ ДОГОВОРА
1.1. Благотворитель безвозмездно передает Организации денежные средства (далее -Пожертвование) на ведение уставной деятельности Организации, ее содержание, а также финансирования программ и проектов Организации.
1.2. Организация принимает пожертвование, поступившее в рамках Договора, для финансирования программ и проектов Организации, направленных на помощь и поддержку детей и взрослых со спинальной мышечной атрофией и другими нервно-мышечными заболеваниями, их семей и близких, а также на оказание помощи и поддержки другим некоммерческим организациям и учреждениям, осуществляющим деятельность, соответствующую целям деятельности Фонда.
1.3. Договор является договором присоединения (ст. 428 Гражданского кодекса РФ). Условия Договора принимаются Благотворителем путем присоединения к настоящему договору в целом. При этом Благотворитель подтверждает, что Договор не содержит обременительных для него условий, которые он не принял бы при наличии у него возможности участвовать в определении условий настоящего Договора.
2. ПОРЯДОК ЗАКЛЮЧЕНИЯ ДОГОВОРА
2.1. Договор считается заключенным в письменной формес момента передачи Благотворителем Организации Пожертвования в порядке, определенном настоящим Договором,что означает безоговорочное принятие всех его условий без каких-либо изъятий или ограничений.
2.5. При перечислении пожертвования в целях идентификации Благотворитель указывает свои контакты: ФИО/наименование юридического лица, адрес электронной почты и/или телефонный номер.
2.6. Пожертвование считается переданными Организации с момента его зачисления на банковский счет Организации.
2.8. Местом заключения Договора является место нахождения Организации.
3. УСЛОВИЯ ДОГОВОРА
3.2. Организация обязана использовать полученное по настоящему Договору Пожертвование исключительно на цели, указанные в п. 1.2. Договора.
3.4. Стороны несут полную ответственность за соблюдение требований Договора, в том числе ответственность о предоставленных сведениях о себе. Каждая из Сторон подтверждает, что она имеет все права и полномочия на заключение Договора и исполнение установленных им обязательств, а также что заключение Договора не нарушает условий иных обязательств Сторон перед третьими лицами.
3.5.Стороны освобождаются от ответственности за неисполнение или ненадлежащее исполнение обязательств по договору, если это неисполнение явилось следствием обстоятельств непреодолимой силы, возникших после заключения договора в результате событий чрезвычайного характера, которые Сторона(ы) не могла(и) ни предвидеть, ни предотвратить разумными мерами (форс-мажора).
3.6. Все споры между Сторонами подлежат рассмотрению в суде по месту нахождения Организации.
3.7. Во всем остальном, что не предусмотрено Договором, Стороны руководствуются действующим законодательством Российской Федерации.
4. РЕКВИЗИТЫ ОРГАНИЗАЦИИ
Юридический адрес 115408, Москва, ул. Борисовские пруды, д. 48 корп. 2 кв. 211
ИНН/КПП 7724342940/772401001
Р/С 40703810438000003439
Кор. Счет 30101810400000000225
БИК 044525225
ОРГН 1157700018816
ОКПО 51247736
Согласие на обработку персональных данных
Пользователь дает согласие на обработку своих персональных данных, как без использования средств автоматизации, так и с их использованием.
Согласие дается на обработку следующих персональных данных (не являющимися специальными или биометрическими):
• фамилия, имя, отчество;
• адрес(а) электронной почты;
• иные данные, предоставляемые Пользователем.
Персональные данные пользователя не являются общедоступными.
3. В ходе обработки с персональными данными будут совершены следующие действия с персональными данными: сбор, запись, систематизация, накопление, хранение, уточнение (обновление, изменение), извлечение, использование, передача (распространение, предоставление, доступ), обезличивание, блокирование, удаление, уничтожение.
4. Передача персональных данных, скрытых для общего просмотра, третьим лицам не осуществляется, за исключением случаев, предусмотренных законодательством Российской Федерации.
5. Пользователь подтверждает, что указанные им персональные данные принадлежат лично ему.
9. Настоящее Согласие является бессрочным, и действует все время до момента прекращения обработки персональных данных, указанных в п.7 и п.8 данного Согласия.
Спинальная мышечная атрофия – одно из самых опасных генетически обусловленных заболеваний, которое обнаруживается у младенцев, подростков, взрослых.

Страшно узнать, что малыш никогда не будет сидеть, стоять, бегать. Еще страшнее видеть, как нормально растущий и развивающийся ребенок вдруг начинает медленно угасать, постоянно падать, через несколько месяцев не может подняться по лестнице, а однажды теряет способность просто встать.
Спинальная мышечная атрофия — что это
Врачи объединяют несколько видов наследственных заболеваний, характеризующихся нарушением движения, в одну группу под названием спинальная мышечная атрофия. В МКБ-10 они идут под кодом G12 с дополнительными указаниями на тип болезни.
Спинальная мышечная атрофия — это разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга.
По данным исследователей, около 0,01-0,02% детей рождаются с диагнозом СМА. Чаще патология встречается у мальчиков и мужчин.
Обнаруживается спинальная мышечная атрофия преимущественно у детей в раннем возрасте. Однако некоторые формы заболевания начинают проявляться только у подростков или уже взрослых людей. Коварство патологии заключается в том, что она постепенно, день за днем отбирает у больных то, что они сумели добиться.
Впервые патологию описал Г. Вердниг. Он обратил внимание на равностороннюю атрофию спинного мозга, его передних рогов, корешков периферических нервов в 1891 г. Уже в следующем году Дж. Хоффман сумел доказать, что речь идет о самостоятельном заболевании. В середине XX в. исследователи Е. Кугелберг и Л. Веландер описали патологию, которая возникает в позднем возрасте и имеет более благоприятный прогноз.
Симптомы
Каждый вид СМА имеет свои особенные признаки, однако существуют некоторые симптомы, которые позволяют объединить разнородные заболевания в одну группу. Это:
- Нарастающая слабость мышц и их атрофия.
- При заболевании, проявившемся после 1-2 лет, заметна деградация уже достигнутых способностей, например, бега, ходьбы.
- Тремор пальцев. Дрожь наблюдается и на языке.
- Деформация скелета.
- Сохранность интеллектуального и психического здоровья у большинства больных.

Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
Наблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.

Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
Причины и механизм развития заболевания
Спинальная амиотрофия развивается из-за мутировавшего SMN гена пятой хромосомы. Если оба родителя – его носители, существует 25%-ная вероятность, что ребенок родится больным.
Мутация гена SMN приводит к нарушению синтеза белка, в результате чего происходит разрушение мотонейронов спинного мозга. Нервные импульсы не проходят к мышцам, которые из-за бездействия атрофируются, человек теряет способность двигаться.
Считается, что теряет работоспособность сначала глубоко расположенная мускульная ткань.
Диагностика
Наиболее точным методом определения спинально-мышечной атрофии у детей является анализ ДНК. Он проводится как у родившегося малыша, так и во время внутриутробного развития. Дополнительно проводятся следующие исследования:

Если у молодых людей, планирующих рождение ребенка, есть родственники с патологией СМА, им рекомендовано пройти генетическую экспертизу.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс

Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Прогноз
То, как будет развиваться болезнь, сколько лет проживет ребенок, зависит от ее типа.
При атрофии типа один прогноз крайне неблагоприятен. Около 50% малышей не доживают и до двух лет. Не больше 10% детей с болезнью Верднига-Гоффмана могут дожить до пяти лет. Причиной гибели чаще всего становится воспаление легких, остановка дыхания, сердца.
Пациенты, которым диагностирована болезнь Дубовица, живут в среднем до 10, иногда 12 лет. Около 30% малышей умирают, не достигнув четырех лет.
При SMA III типа детская смертность встречается реже. У многих пациентов симптомы появляются в предподростковом-подростковом возрасте. Через несколько лет они перестают ходить. Далее, по нарастающей, отмечается атрофия мышц внутренних органов, в том числе дыхательных.
Считается, что заболевание IV типа не влияет на продолжительность жизни, тем не менее, оно ведет к инвалидизации.
Профилактика
Мер, направленных на профилактику и предотвращение развития СМА, не существует. Женщина, ожидающая рождения ребенка, может заподозрить проблему, обратив внимание на слабость шевелений плода. Проведенный ДНК-анализ может подтвердить или развеять подозрения. При необходимости проводится медицинская комиссия, которая может порекомендовать прерывание беременности. Врач обязательно рассказывает о заболевании, его течении и последствиях.
После диагностики заболевания у уже родившегося ребенка его окружают заботой и вниманием. Использование системы искусственной вентиляции легких, отсасывателей мокроты, специальных приспособлений для движения малыша, который может передвигаться, помогают улучшить качество жизни и помочь ребенку жить. Рекомендовано регулярно делать массаж, физиопроцедуры. Детей даже с ограниченными движениями возят в бассейн.
Спинальная амиотрофия – опасная, пока не поддающаяся лечению патология. Она характеризуется атрофией мышц. Возникает в разном возрасте. Прогноз в большинстве случаев неблагоприятный.
Для подготовки статьи использовались следующие источники:
Селиверстов Ю. А., Клюшников С. А., Иллариошкин С. Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Журнал Нервные болезни — 2015
Лепесова М. М., Ушакова Т. С., Мырзалиева Б. Д. Дифференциальная диагностика спинальной мышечной амиотрофии первого типа // Вестник Алматинского государственного института усовершенствования врачей — 2016
Читайте также: