
Спинальная мышечная атрофия фонды



С 2015 года наш фонд помогает больным СМА и их близким, не только оставаясь с ними в постоянном контакте, но и развивая системные проекты, которые дают возможность с каждым днем увеличивать объем помощи.
Деятельность фонда направлена на обучение специалистов и родителей, распространение знаний и повышение уровня информированности о заболевании.
Мы постоянно на связи с ведущими российскими и мировыми экспертами по СМА, регулярно переводим на русский язык и издаем информационные материалы, исследования и новости о заболевании и терапии.
Мы всегда рады поделиться собственным опытом и наработками не только с подопечными, но и с другими некоммерческими организациями и медицинскими учреждениями.

Друзья, сделайте подарок нашему фонду и всем семьям, в которых живут больные Спинальной мышечной атрофией - подпишите петицию. У всех больных должна быть реальная возможность получить доступ к терапии!
Сегодня нам нужна ваша поддержка!




- Записи сообщества
- Поиск
Друзья, мы боремся за каждого ребёнка- СМА, но это невозможно без нашего участия в Международной ассоциации, располагающей последними данными о том, чем и как можно помочь нашим детям.
Показать полностью… До закрытия сбора осталось собрать 25 тысяч, и мы, оплатив участие, сможем дальше вам помогать! Сможем ли собрать эту сумму быстро? Нам кажется, это возможно, ведь основная сумма была собрана в течение нескольких дней, потому что все сплотились и поддержали друг друга.
К нам обращаются СМА-пациенты, потому что мы профессионалы. Чтобы быть профессионалом и оказывать действенную помощь, мы должны владеть передовыми знаниями в области редкого заболевания, и знать все о СМА не только в России, но и в Европе, в мире.
Есть организация СМА Европа (Европейская ассоциация европейских организаций) , участники которой делятся между собой мировым опытом, открытиями, новейшими разработками Нам нельзя отставать, чтобы нашим пациентам было доступно все лучшее, что придумано для облегчения жизни со СМА. Кто-то только начинает жизнь со СМА, кто-то прошёл долгий путь, что-то узнал, придумал, открыл. Мы должны быть вместе! Вне зависимости от расстояния, все, кто занимается проблемами СМА должны быть в одной команде и иметь возможность поддержать друг друга, подхватывая и передавая взаимный положительный опыт и избегая ошибок, уже зная, где они могут быть, чтобы каждая семья-СМА не проходила свой личный трудный путь, определенные сложности которого уже научились преодолевать в других странах.
Мы на защите наших пациентов и хотим внедрять успешные практики в своей стране. О людях-СМА, несмотря на редкость заболевания, мир должен знать, слышать их, для этого нужно объединяться и становиться практически семьей. В ассоциации СМА Европа нет французов, русских, поляков и англичан, есть люди, объединённые желанием помочь детям и взрослым с непростым редким диагнозом. Успехи такой объединённой общей целью деятельности говорят сами за себя.
Каждая организация, которая вступает в ассоциацию СМА Европа должна ежегодно оплачивать членский взнос в сумме 1500 евро. Эти средства идут на поддержание работы ассоциации, разработки передовых инициатив, которые мы обязательно сможем применять для наших детей с диагнозом СМА. И мы просим вашей поддержки для оплаты нашего членского взноса. Мы не можем остаться за кормой мировых достижений в области СМА. За каждыми оптимистичными цифрами, показателями — судьбы людей, которым постепенно становится легче жить. Мы хотим продолжать это делать.
У одного из 6-10 тысяч детей есть мутация в гене, которая со временем не даст ему двигаться, есть и даже дышать. Но ребенок может вести нормальную жизнь, если вовремя получит терапию, желательно до того, как появятся первые признаки спинальной мышечной атрофии (СМА).
Первое лекарство для пациентов со СМА появилось всего несколько лет назад и доходит до российских пациентов с трудом. Сведения о новых средствах одновременно противоречивы и полны надежды.

Фото: Sebastian Gollnow/dpa/picture-alliance
Что такое СМА
Спинальная мышечная атрофия (СМА) — это неизлечимое заболевание, при котором у человека поврежден или вовсе отсутствует ген, отвечающий за работу двигательных нейронов. Болезнь приводит к поражению нервной системы и постепенной атрофии мышц. В результате у человека сильно искривляется позвоночник, ему становится трудно дышать, он не может двигаться, при этом его интеллект полностью сохранен.
СМА может проявиться как в первые месяцы жизни, так и в возрасте от полутора до двух лет. Но чем раньше диагностировать заболевание, тем лучше для ребенка, — лечение наиболее эффективно, когда симптомы еще не проявились. Поэтому эксперты настаивают на том, что всем новорожденным нужно проводить скрининг на СМА. Болезнь неизлечима, но если человек вовремя получает лекарства и специальное медицинское оборудование, то может полноценно жить.
Спинраза
Рисдиплам
Золгенсма
Однако эксперты относятся к результатам с осторожностью: наблюдения за участниками испытаний длятся не больше четыре лет. Кроме того, в тестированиях принимали участие только несколько десятков детей со СМА в возрасте около семи месяцев, тогда как компания рекомендует использовать препарат детям до двух лет.
Крик отчаяния
Закрыть сбор на препарат для Кати помог анонимный благотворитель, который пожертвовал на лечение девочки 145 миллионов рублей. Сейчас надеются собрать средства на лечение семьи из Зеленограда, Калининграда, Мурманска, Воронежа, Москвы, Красноярска, Екатеринбурга, Владикавказа, Югорска и Воронежа.
Каждый день мы пишем о самых важных проблемах в нашей стране. Мы уверены, что их можно преодолеть, только рассказывая о том, что происходит на самом деле. Поэтому мы посылаем корреспондентов в командировки, публикуем репортажи и интервью, фотоистории и экспертные мнения. Мы собираем деньги для множества фондов — и не берем из них никакого процента на свою работу.
Пожалуйста, подпишитесь на любое пожертвование в нашу пользу. Спасибо.

На Ваш почтовый ящик отправлено сообщение, содержащее ссылку для подтверждения правильности адреса. Пожалуйста, перейдите по ссылке для завершения подписки.
Исключительные права на фото- и иные материалы принадлежат авторам. Любое размещение материалов на сторонних ресурсах необходимо согласовывать с правообладателями.
По всем вопросам обращайтесь на mne@nuzhnapomosh.ru
Нашли опечатку? Выделите слово и нажмите Ctrl+Enter
- О фонде
- Контакты
- Отчеты
- Для НКО
- Персональные данные
- Пожертвовать
- Стать волонтером
- Частые вопросы
- ВКонтакте
- Telegram
- Youtube
- Дзен
Нашли опечатку? Выделите слово и нажмите Ctrl+Enter
(Протокол № 1 от 20.01.2020 г.)
Благотворительный фонд помощи социально-незащищенным гражданам "Нужна помощь"
Адрес: 119270, г. Москва, Лужнецкая набережная, д. 2/4, стр. 16, помещение 405
ИНН: 9710001171
КПП: 770401001
ОГРН: 1157700014053
Номер счета получателя платежа: 40703810238000002575
Номер корр. счета банка получателя платежа: 30101810400000000225
Наименование банка получателя платежа: ПАО СБЕРБАНК РОССИИ г. Москва
БИК: 044525225
Персональные данные обрабатываются Фондом для целей исполнения договора пожертвования, заключенного между Вами и Фондом, для целей направления Вам информационных сообщений в виде рассылки по электронной почте, СМС-сообщений. В том числе (но не ограничиваясь) Фонд может направлять Вам уведомления о пожертвованиях, новости и отчеты о работе Фонда. Также Персональные данные могут обрабатываться для целей корректной работы Личного кабинета пользователя Сайта по адресу my.nuzhnapomosh.ru.
Персональные данные будут обрабатываться Фондом путем сбора Персональных данных, их записи, систематизации, накопления, хранения, уточнения (обновления, изменения), извлечения, использования, удаления и уничтожения (как с использованием средств автоматизации, так и без их использования).
Передача Персональных данных третьим лицам может быть осуществлена исключительно по основаниям, предусмотренным законодательством Российской Федерации.
Персональные данные будут обрабатываться Фондом до достижения цели обработки, указанной выше, а после будут обезличены или уничтожены, как того требует применимое законодательство Российской Федерации.
Что говорят специалисты о новом лекарстве от СМА


Русфонд получает много просьб помочь с покупкой золгенсмы – препарата для генной терапии спинальной мышечной атрофии (СМА). Родители тяжелобольных детей надеются, что новое лекарство может остановить развитие болезни. На деле это не так, говорят и зарубежные, и российские специалисты.
Один из крупнейших экспертов по СМА в Европе профессор Лоран Сервэ утверждает, что золгенсма эффективна только для младенцев в возрасте до семи месяцев и не может вылечить детей, у которых уже проявились симптомы СМА. В Валлонском регионе Бельгии, где работает Сервэ, СМА входит в обязательную программу скрининга новорожденных. Это позволяет выявлять генетическое заболевание еще до появления симптомов.
Беспрецедентная цена золгенсмы – свыше $2 млн за инъекцию – оправданна лишь в случае, если препарат дают младенцу до появления симптомов, заявил Сервэ в интервью бельгийскому изданию La Libre.
Островская обратила внимание на еще один важный момент. Даже собрав средства на покупку лекарства, родители столкнутся с проблемой его применения:
– Родителей тяжелобольных детей можно понять – они хватаются за любую возможность помочь своему ребенку. Ну допустим, они соберут эти деньги, купят лекарство, но как и где они его будут вводить? У себя дома? Ни одна больница не будет применять не зарегистрированный в России препарат, если его используют не в рамках какого-то специального исследования. Для этого нужно специальное согласование, оформление – в целом очень сложный процесс.
Наблюдения за применением золгенсмы ведутся всего четыре года на небольшой группе из 25 пациентов, отмечает Светлана Артемьева, заведующая психоневрологическим отделением № 2 Научно-исследовательского клинического института педиатрии имени академика Ю.Е. Вельтищева (Москва):
– В том, что препарат эффективен, сомневаться не приходится, но хватит ли одной инъекции на всю жизнь? Этот вопрос еще обсуждается учеными. Может быть, в дальнейшем пациенту потребуется поддержка другими препаратами, тем же рисдипламом или спинразой, потому что у них разные точки приложения. Клинические исследования действительно показывают, что все препараты, не только золгенсма, доказывают свою наибольшую эффективность, когда их начинают применять на ранней, досимптомной стадии СМА. Если лечить детей до шести месяцев, то действительно можно остановить прогрессирование болезни и ребенок будет развиваться соответственно возрасту. Но если симптомы уже есть и ребенок потерял какие-то двигательные функции, то ситуация иная. Это можно сравнить с инсультом: в зависимости от тяжести поражения некоторые пациенты восстанавливаются полностью, у других сохраняются минимальные симптомы, а у некоторых – тяжелые двигательные нарушения.
Скрининг новорожденных в России пока находится в стадии пилотного проекта. Если он окажется удачным, генетики будут добиваться введения его по всей стране, сообщает Артемьева.
Александр Поляков, профессор РАН, ведущий специалист в области молекулярной генетики и ДНК-диагностики наследственных заболеваний, заведующий лабораторией ДНК-диагностики Медико-генетического научного центра имени академика Н.П. Бочкова (Москва), отметил, что доступных данных об эффективности золгенсмы на конференциях и в печати практически нет. Спинраза изучена лучше:
– Механизмы действия золгенсмы и спинразы различны, но оба препарата направлены не на следствия, а на устранение причины болезни. Разница в цене есть, но нужно помнить, что спинразу требуется вводить многократно, в отличие от золгенсмы. Вообще, я считаю, что цена на определенный препарат – это вопрос переговоров между фармкомпанией и государством и она должна быть тем ниже, чем больше в стране больных, получающих данный препарат, – добавил Поляков. – Что касается возраста пациентов, то, безусловно, любой препарат наиболее эффективен на самых ранних стадиях болезни. А эти стадии различны для разных типов СМА.
Германенко отмечает, что золгенсму сопровождает агрессивный пиар, который действует на родителей пациентов:
– Не надо принимать желаемое за действительное. Но это очень трудно для родителей больных детей. Вся шумиха приводит к тому, что золгенсма воспринимается как панацея, как лекарство, которое может излечить ребенка независимо от того, на какой стадии заболевания он находится. Однако это не так. Все три лекарства – и золгенсма, и спинраза, и рисдиплам – препараты для терапии, а не для излечения. СМА пока еще неизлечимое заболевание, это надо признать. Но чем раньше мы начинаем лечение, тем эффективнее будет любой препарат из имеющихся на сегодняшний день. При этом у нас сейчас большие сложности с доступом пациентов даже к зарегистрированному препарату – спинразе, и это действительно серьезная проблема. Золгенсма не зарегистрирована в России. И тут нужно отметить, что незарегистрированные препараты всегда должны рассматриваться как препараты второй линии. Кому-то покажется, что это нечестно или несправедливо. Но так работают медицинские системы во всем мире, и такие критерии были введены неслучайно. О золгенсме можно сказать так: ожидания от этого препарата на сегодняшний день выше, чем возможности самого препарата.
Что такое спинальная мышечная атрофия?

Спинальная мышечная атрофия (СМА) – это генетическое заболевание, поражающее часть нервной системы, которая контролирует сознательное движение мышц.
Большинство нервных клеток, которые контролируют мышцы, расположены в спинном мозге, из-за чего заболевание и получило своё название – “спинальная”. СМА является мышечной потому, что заболевание в основном влияет на мышцы, которые не получают сигналы от нервных клеток. Атрофия – это медицинский термин для “уменьшения”, что и происходит с мышцами, когда они не активны.
При СМА происходит утрата нервных клеток, называемых двигательными (моторными) нейронами, в спинном мозге. СМА классифицируется как болезнь двигательных нейронов.
В самой распространённой форме СМА (дефектный ген на хромосоме 5, или SMN-связанная СМА) существует широкая градация в возрасте начала заболевания, симптомах и скорости прогрессирования. Эту форму часто подразделяют на четыре типа СМА (с 1 по 4).
Возраст, при котором симптомы СМА начинают проявляться, примерно коррелирует со степенью поражения двигательной функции. Чем раньше проявилась СМА, тем большее влияние оказывается на двигательную функцию. Дети, у которых симптомы появляются при рождении или в младенчестве, как правило, имеют самый низкий уровень двигательной способности (тип 1). СМА 2 и 3 типа проявляется в детском возрасте, а СМА 4 типа – у подростков или взрослых. При СМА со второго по четвёртый тип уровень двигательной активности гораздо выше, чем при СМА 1 типа.
Что вызывает СМА?
СМА, возникающая из-за мутации на 5-й хромосоме, вызвана дефицитом белка под названием SMN (survival of motor neuron – выживаемость мотонейронов). Этот белок, как следует из его названия, необходим для нормальной функции двигательных нейронов. Его недостаток вызван генетическим дефектом (мутацией) на 5-й хромосоме в гене под названием SMN1. Соседние гены SMN2 могут частично компенсировать нефункциональные гены SMN1.
Другие редкие формы СМА (не связанные с мутациями на 5-й хромосоме) вызваны мутациями в других генах.
Симптомы СМА
СМА варьируется от средней до тяжёлой формы. Мышцы, расположенные ближе к центру тела (проксимальные мышцы), как правило, повреждены в большей степени, чем мышцы, расположенные дальше от центра (дистальные мышцы).
Основным симптомом СМА, вызванной мутацией на 5-й хромосоме из-за дефекта в гене SMN, является слабость сокращающихся мышц. Больше всего страдают мышцы, расположенные ближе к центру тела, такие как мышцы плеч, бедер и верхней части спины. Особые трудности возникают, если повреждаются мышцы, используемые для дыхания и глотания. При ослаблении мышц спины может появиться искривление позвоночника.
При СМА, вызванной мутацией на 5-й хромосоме, существует широкая градация в возрасте начала заболевания и степени двигательной активности. Эти показатели относительно зависят от количества функционального белка SMN в организме, что в свою очередь зависит от количества копий гена SMN2.
При СМА, вызванной мутацией на 5-й хромосоме, чувствительное, умственное и эмоциональное развитие совершенно нормально.
Некоторые формы СМА не связаны с 5-й хромосомой или дефицитом белка SMN. Эти формы сильно различаются по степени тяжести. Большинство форм СМА, в частности, связанных с 5-й хромосомой, в основном влияют на проксимальные мышцы, но некоторые другие формы поражают в основном дистальные мышцы (расположенные дальше от центра тела) – по крайней мере, в начале.
Прогрессирование СМА
При формах СМА, связанных с 5-й хромосомой, чем позднее начинаются симптомы и чем больше в организме вырабатывается белка SMN, тем более мягким будет ход заболевания. В прошлом у детей с СМА, как правило, продолжительность жизни была не больше двух лет, однако в настоящее время большинство врачей считают, что формы СМА, вызванные нехваткой белка SMN, могут быть контролируемыми, и предпочитают не делать жёсткие прогнозы относительно ожидаемой продолжительности жизни или слабости, основываясь исключительно на возрасте начала заболевания.
Каково состояние исследований по СМА?
Исследования сосредоточены на разработке стратегий по увеличению производства организмом белка SMN, который отсутствует или вырабатывается в малых количествах при СМА, связанной с мутацией на 5-й хромосоме. Кроме того, исследования для этой формы СМА, а также для других форм, включают методы, которые могут помочь двигательным нейронам выживать в неблагоприятных условиях.
Диагностика спинальной мышечной атрофии
- Общий список клиник и врачей, занимающихся нервно-мышечными заболеваниями, — по ссылке.
- Российский благотворительный фонд помощи больным СМА – “Семьи СМА”, который является единственным фондом поддержки пациентов с СМА и их семей. Фонд разрабатывает информационные материалы, помогает с обеспечением медицинским оборудованием и лекарствами, проводит обучающие семинары и встречи. При фонде действует Клиника СМА.
Проект “Клиника СМА” проводится совместно с специалистами разного профиля, имеющими опыт наблюдения и работы с пациентами со спинальной мышечной атрофией.
| Сегодня фонд “Семьи СМА” существует благодаря пожертвованиям. Для помощи фонду вы можете оформить разовое или регулярное пожертвование: | Пожертвовать |
Что важно знать при СМА?
Важно! Регистры (реестры) пациентов со спинальной мышечной атрофией
По мере того, как разрабатываются новые препараты, появляется необходимость их тестирования в клинических условиях, и иногда требуются годы, чтобы найти необходимое количество пациентов для исследований, поскольку спинальная мышечная атрофия является редким (орфанным) заболеванием.
Для этого в разных странах ведутся реестры пациентов с СМА — базы данных по генетической и клинической информации о людях, страдающих СМА и желающих ускорить процесс исследований. Реестр позволяет специалистам получить информацию о состоянии и количестве больных данным заболеванием. Данная информация способствует развитию и улучшению стандартов лечения пациентов. Он используется, чтобы найти участников для проведения клинических испытаний, а также помочь специалистам получить больше информации о заболевании.
Организации и сообщества, посвящённые спинальной мышечной атрофии
Контакты фонда: телефон – +7 495 544 49 89, почта – info@f-sma.ru.
Месяц осведомлённости о спинальной мышечной атрофии

Разместите у себя на страничке в социальной сети фото с эмблемой акции на срок, который будет для Вас оптимальным. Так вы поможете рассказать об этом нервно-мышечном генетическом заболевании. Вместе мы можем сделать больше!
Техническая подсказка: приложение создает альбом Twibbon Photos в разделе “Ваши фотографии” на Фейсбук, если захотите установить фото с эмблемой в качестве фотографии профиля, откройте фото и в левом нижнем углу нажмите “сделать фото профиля”. Или сохраните изображение на свой компьютер и сможете ставить картинку профиля на любом другом ресурсе.

Частота заболевания
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.
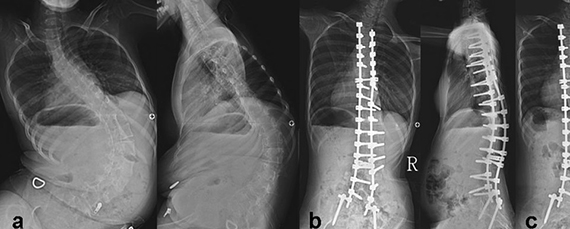
От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III, болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
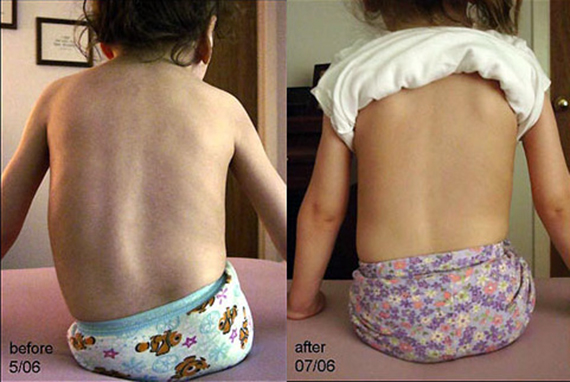
На сегодняшний момент радикального лечения от СМА не существует.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300.
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Сколько больных СМА в России?
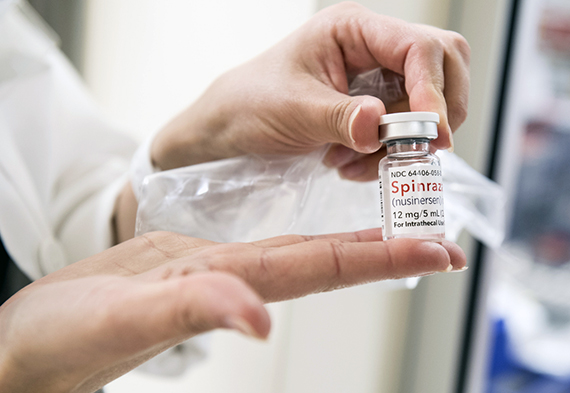
Кто в России помогает людям со СМА и их семьям
Известные люди со СМА

Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
Программист из Владимира Валерий Спиридонов. Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Читайте также: