Болезнь Стилла: причины, симптомы и лечение
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Болезнь Стилла — тяжелое заболевание, проявляющееся лихорадкой, полиартритом. Преходящими высыпаниями на коже и системным воспалительным поражением соматических органов. Болезнь Стилла диагностируется с применением методики исключения других заболеваний на основании клинических симптомов, лабораторных данных, результатов исследования пораженных суставов, лимфоретикулярной и сердечно-легочной системы. Лечение болезни Стилла проводят в основном нестероидными противовоспалительными и глюкокортикоидами, препаратами резерва являются цитостатики.

Общие сведения
Болезнь Стилла была описана еще в 1897 году британским врачом Джорджем Стиллом. Долгое время она считалась тяжелой формой ювенильной формы ревматоидного артрита. Лишь в 1971 году Эриком Байуотерсом были опубликованы многочисленные наблюдения этого заболевания у взрослых пациентов. Согласно статистике, которую приводит современная мировая ревматология, распространенность болезни Стилла в последнее время составляет примерно 1 человек на 100 тыс. населения. Лица женского и мужского пола одинаково подвержены заболеваемости. Наибольшее число случаев болезни Стилла приходится на детей в возрасте до 16 лет.
Из-за отсутствия специфических симптомов заболевания, пациентам с болезнью Стилла зачастую, несмотря на отрицательные результаты бактериологических посевов крови, ставят диагноз «сепсис», по поводу которого они проходят неоднократные курсы антибиотикотерапии. Отмечено, что около 5% случаев болезни Стилла первоначально трактуются врачами как «лихорадка неясного генеза».

Причины возникновения болезни Стилла
Многочисленные исследования в области этиологии болезни Стилла так и не дали ответа на вопрос о ее причинах. Внезапное начало, высокая лихорадка, лимфоаденопатия и лейкоцитоз крови указывают на инфекционный характер заболевания. Однако единый возбудитель пока не выявлен. В отдельных случаях болезни Стилла у пациентов был выявлен вирус краснухи, в других — цитомегаловирус. Наблюдались случаи заболевания ассоциированные с вирусом парагриппа, вирусом Эпштейна-Барра, микоплазмой, эшерихиями.
Нельзя исключить наличие наследственной предрасположенности к развитию болезни Стилла. Но окончательные результаты, подтверждающие связь заболевания с локусами HLA, пока не получены. Иммунологическая теория, относящая болезнь Стилла к аутоиммунным заболеваниям, подтверждается лишь в некоторых случаях, когда у больных обнаруживаются ЦИК, обуславливающие развитие аллергического васкулита.
Симптомы болезни Стилла
Лихорадка при болезни Стилла отличается подъемом температуры до высоких цифр (39°С и выше). В отличие от большинства инфекционных заболеваний она не является постоянной. Наиболее характерен одноразовый подъем температуры в течение суток, обычно в вечернее время. Реже наблюдаются 2 температурных пика за сутки. У большинства больных температура между пиками снижается до нормальных цифр, что сопровождается значительным улучшением общего состояния. Примерно у 20% пациентов с болезнью Стилла нормализации температуры тела не происходит.
Высыпания при болезни Стилла, как правило, возникают на высоте подъема температуры тела и носят приходящий характер: то исчезают, то появляются вновь. Элементы сыпи представлены в основном плоскими розовыми пятнами (макулами) или папулами, расположенными в проксимальных отделах конечностей и на туловище, реже — на лице. В 30% случаев болезни Стилла высыпания возвышаются над общей поверхностью кожного покрова и возникают в местах травмирования или сдавления кожи (феномен Кебнера). Иногда они сопровождаются зудом. Розовый цвет сыпи, ее периодическое исчезновение и отсутствие субъективных ощущений часто делают высыпания незаметными для пациентов. В некоторых случаях для выявлении сыпи врачу приходится осматривать пациента сразу после теплого душа или прибегать к тепловому воздействию на кожу, например, путем наложения теплых салфеток. Встречаются атипичные кожные проявления болезни Стилла: петехиальные кровоизлияния, узловатая эритема, алопеция.
Суставной синдром. Артралгии, наряду с миалгиями, в начале болезни Стилла относят к общим проявлениям заболевания, обусловленным высоким подъемом температуры. На начальном этапе артрит может поражать лишь один сустав. Затем поражение принимает характер полиартрита с вовлечением голеностопных, коленных, лучезапястных, локтевых, тазобедренных, височно-челюстных, межфаланговых, плюсне-фаланговых суставов. Наиболее типичным для болезни Стилла является развитие артрита межфаланговых дистальных суставов кисти. Эта особенность позволяет дифференцировать заболевание от ревматоидного артрита, ревматической лихорадки, системной красной волчанки, для которых не характерно поражение этих суставов в молодом возрасте.
Поражение лимфоретикулярных органов включает гепатоспленомегалию и лимфаденопатию. У 65% заболевших болезнью Стилла наблюдается лимфаденит. В половине случаев заболевания наблюдается увеличение шейных лимфоузлов. Увеличенные лимфоузлы при болезни Стилла сохраняют свою подвижность, имеют умеренно плотную консистенцию. Выраженное уплотнение лимфатического узла, его изолированное увеличение или спаянность с окружающими тканями должны настораживать в онкологическом плане. В атипичных случаях лимфаденит может принимать некротический характер.
Боль в горле беспокоит 70% пациентов с болезнью Стилла и проявляется обычно в начале заболевания. Она характеризуется выраженным жжением в горле и носит постоянный характер.
Сердечно-легочные проявления болезни Стилла наиболее часто носят характер серозита: плеврита и/или перикардита. В 20% случаев наблюдается асептический пневмонит, зачастую протекающий с симптомами двусторонней пневмонии (кашель, одышка, высокая температура), которые не проходят на фоне интенсивной антибиотикотерапии. К более редким поражениям, встречающимся при болезни Стилла, относятся: миокардит, тампонада сердца, появление клапанных вегетаций с клинической картиной инфекционного эндокардита, респираторный дистресс-синдром.
Диагностика болезни Стилла
Отсутствие специфических диагностических признаков болезни Стилла делают ее диагностику затруднительной для ревматолога, требующей определенного периода наблюдения пациента и часто основанной на исключении других заболеваний. В клиническом анализе крови отмечается выраженный лейкоцитоз и ускоренное СОЭ. У подавляющего большинства пациентов с болезнью Стилла СОЭ выше 50 мм/ч. В биохимическом анализе крови выявляется повышенный уровень белков, характерных для острой воспалительной фазы: СРБ, ферритина, сывороточного амилоида А. При этом, не смотря на типичные для болезни Стилла клинические признаки выраженного системного воспаления, в крови не обнаруживаются ревматоидный и антинуклеарный фактор, а бакпосев крови на стерильность дает отрицательный результат. Биохимические пробы печени показывают увеличение активности ее ферментов.
Рентгенологическое исследование суставов выявляет выпот в полости сустава, отечность мягких тканей, реже — остеопороз образующих сустав костей. У пациентов с хронической формой болезни Стилла типичным является наличие анкилозов в суставах запястья. При проведении пункции суставов получают асептическую синовиальную жидкость с воспалительными изменениями.
При необходимости пациентам с болезнью Стилла проводится биопсия лимфатического узла, позволяющая исключить его злокачественное метастатическое поражение. Сердечно-легочные проявления болезни Стилла требуют консультации кардиолога и пульмонолога, проведения рентгенографии легких, УЗИ плевральной полости, ЭКГ, УЗИ сердца и т. п. Дифференциальный диагноз болезни Стилла проводят с ревматоидным артритом, псориатическим артритом, дерматомиозитом, лимфомой, туберкулезом, саркоидозом, гранулематозным гепатитом, инфекционным эндокардитом, системными васкулитами и др.
Лечение болезни Стилла
В остром периоде для 25% пациентов достаточно назначения препаратов из группы нестероидных противовоспалительных. Их прием в зависимости от клиники болезни Стилла занимает от 1 до 3 месяцев. Изменения со стороны сердца и легких являются показанием к глюкокортикостероидной терапии препаратами преднизолона или дексаметазона. Однако эти препараты не всегда оказывают достаточный эффект. При хроническом течении болезни Стилла для уменьшения дозы кортикостероидов может применяться метотрексат. Препаратом резерва для пациентов с тяжелыми формами заболевания может являться циклофосфамид. В отдельных, резистентных к традиционному лечению случаях болезни Стилла, возможно применение инфликсимаба и этанерцепта.
Прогноз болезни Стилла
Исходом болезни Стилла может быть спонтанное выздоровление, переход в рецидивирующую или хроническую форму. Выздоровление наступает у 1/3 больных, обычно в течение 6-9 месяцев от начала заболевания. Рецидивирующее течение болезни Стилла у 2/3 пациентов характеризуется возникновением лишь одной атаки (обострения) заболевания, которая может случиться в период от 10 мес до 10 лет. У незначительной части пациентов наблюдается циклическое рецидивирующее течение заболевания с повторными атаками. Наиболее тяжелой является хроническая форма болезни Стилла, протекающая с выраженным полиартритом, приводящим к ограничению движений в суставах. Причем, ранее появление симптомов артрита является неблагоприятным прогностическим признаком.
Среди взрослых пациентов с болезнью Стилла пятилетняя выживаемость сравнима с таковой при СКВ и составляет 90-95%. Больные могут погибнуть от вторичной инфекции, амилоидоза, печеночной недостаточности, нарушений свертывания, сердечной недостаточности, туберкулеза легких, респираторного дистресс-синдрома.
Прогрессирующий надъядерный паралич ( Прогрессирующая надъядерная офтальмоплегия , Синдром Стила-Ричардсона-Ольшевского )
Прогрессирующий надъядерный паралич — это дегенеративное церебральное заболевание с преимущественным поражением среднего мозга, ядерно-корковых путей, подкорковых образований. Составляющими клинической картины выступают акинетико-ригидная форма паркинсонизма, атаксия, офтальмоплегия, когнитивное снижение, псевдобульбарный синдром. Диагностика осуществляется по клиническим данным, результатам церебральной МРТ и цереброваскулярных исследований. В терапии препаратами выбора являются леводопа, мемантин, антидепрессанты из группы ингибиторов обратного захвата серотонина.
МКБ-10

Прогрессирующий надъядерный паралич (ПНП) — дегенеративное поражение головного мозга неясной этиологии. Наряду с болезнью Альцгеймера, мультисистемной атрофией, кортикобазальной дегенерацией, болезнью Пика, ПНП относится к таупатиям, характеризующимся образованием включений тау-протеина в нейронах и глиальных клетках. Прогрессирующий надъядерный паралич впервые был подробно описан в 1963-64 годах канадскими неврологами Стилом и Ричардсоном в соавторстве с патоморфологом Ольшевским, в честь которых носит название синдром Стила-Ричардсона-Ольшевского. Распространённость заболевания согласно различным информационным источникам варьирует в пределах 1,4-6,4 случая на 100 тыс. населения. Манифестация клинической симптоматики приходится на возрастной период от 55 до 70 лет, с возрастом вероятность развития заболевания увеличивается. Лица мужского пола в большей степени подвержены болезни по сравнению с женщинами.

Причины ПНП
Этиофакторы, запускающие дегенеративные процессы определённой церебральной локализации, остаются неизвестными. Большинство случаев болезни имеют спорадический характер. Отдельные семейные варианты с предположительным аутосомно-доминантным наследованием были выявлены после 1995 года. Молекулярно-генетические исследования показали, что некоторые формы ПНП обусловлены дефектами кодирующего тау-белок гена, локализованного в локусе 17q21.31. Наиболее вероятным представляется мультифакторный механизм возникновения патологии, реализующийся на фоне генетической предрасположенности.
Патогенез
Ведущим патогенетическим механизмом считается дисметаболизм церебральных внутриклеточных белков, сопровождающийся избирательной агрегацией отдельных белков (тау-протеина, убиквитина) в определённых группах мозговых клеток. Патологические включения нарушают жизнедеятельность нейронов, запускают процесс деградации и запрограммированной гибели (апоптоза). Дегенеративные изменения носят селективный характер, распространяются преимущественно на средний мозг, зубчатые мозжечковые ядра и подкорковые структуры: черную субстанцию, бледный шар, таламус, ретикулярную формацию, субталамическое ядро. В меньшей степени поражается кора префронтальных и височных зон.
Патоморфологическая картина ПНП представлена наличием нейрофибриллярных клубочков, глиальных включений, нитевидных белковых образований в нейронах указанных церебральных структур. Макроскопически определяется атрофия среднего мозга с существенным уменьшением его сагиттального размера. Поражение среднего мозга обуславливает надъядерный паралич глазодвигательной мускулатуры, дегенерация кортико-бульбарных трактов — псевдобульбарные проявления. Нейрохимические исследования выявляют пониженную концентрацию дофамина в стриатуме, лежащую в основе паркинсонического симптомокомплекса.
Симптомы ПНП
Прогрессирующий надъядерный паралич характеризуется неспецифичным клиническим дебютом. Симптоматика этого периода представлена непривычной утомляемостью, сниженной работоспособностью, цефалгиями, головокружением, пониженным настроением, сужением круга интересов, нарушениями сна, включающими бессонницу ночью и гиперсомнию днём. В последующем присоединяются симптомы акинетико-ригидного паркинсонизма. Постуральный тремор у большинства пациентов отсутствует. Мышечная ригидность выражена преимущественно в аксиальной мускулатуре — мышцах, идущих вдоль шейного отдела позвоночника, соединяющих его с черепом. Больные жалуются на скованность в шее, спине. Повышение тонуса в задних мышцах шеи приводит к типичному «горделивому» положению головы пациента. Характерна паркинсоническая атаксия, обусловленная расстройством координации положения туловища и нижних конечностей относительно центра тяжести. Затруднения в поддержании равновесия в процессе ходьбы приводят к частым падениям назад.
Отличительной особенностью ПНП выступает офтальмоплегия, возникающая в среднем спустя 2-3 года от дебюта заболевания. На фоне замедленного движения глазных яблок происходит паралич взора в вертикальной плоскости, пациент не может опустить глаза вниз. Из-за редкого моргания больной ощущает дискомфорт, жжение в глазах. Возможны расплывчатость зрения, расстройство конвергенции, блефароспазм. Прогрессирующий надъядерный офтальмопарез сопровождается ограничением взора вниз и вверх, со временем может приводить к глазодвигательным нарушениям в горизонтальной плоскости. При развитии полной офтальмоплегии формируется ретракция верхних век, что придаёт лицу удивлённое выражение.
В клинической картине ПНП относительно рано возникают псевдобульбарные проявления: дизартрия, дисфагия, насильственный плач или смех. Происходят изменения личностно-эмоциональной сферы, больные становятся замкнутыми, апатичными, демотивированными, безразличными. Когнитивные нарушения в большинстве случаев присоединяются в разгаре болезни, в 10-30% случаев — на стадии дебюта. Характерно интеллектуальное снижение, расстройства абстрактного мышления и памяти, зрительно-пространственная апраксия, элементы агнозии. Деменция наблюдается у 60% пациентов с 3-летним стажем заболевания.
Осложнения
В начальном периоде падения больного без возможности скоординировать свои движения приводят к ушибам и переломам. Спустя несколько лет прогрессирующий олигобрадикинетический синдром приковывает пациентов к постели. При отсутствии должного ухода обездвиженность опасна развитием контрактур суставов, пролежней, застойной пневмонии. Прогрессирующий псевдобульбарный паралич обуславливает попёрхивание пищей с риском асфиксии, аспирационной пневмонии. Ночные апноэ могут стать причиной внезапной смерти во сне. Серьёзным осложнением является присоединение интеркуррентных инфекций (пневмонии, цистита, пиелонефрита), поскольку на фоне сниженного иммунитета существует высокий риск развития сепсиса.
Диагностика
Вероятными ранними критериями ПНП являются начало после 40-летнего возраста, прогрессирующий характер, парез горизонтального взора, выраженная постуральная неустойчивость с эпизодами падений. Постановка достоверного диагноза возможна при наличии гистологически подтверждённых патогномоничных для ПНП изменений в тканях мозга. Перечень необходимых диагностических исследований включает:
- Осмотр невролога. В неврологическом статусе ведущим синдромом является симметричная олигобрадикинезия. Наблюдается гипомимия, ретроколлис (патологическая установка шеи), парез вертикального взора, симптомы орального автоматизма, повышение сухожильных рефлексов. Выражена постуральная неустойчивость.
- Нейропсихологическое тестирование. Проводится психиатром, нейропсихологом с использованием специальных тестов, заданий (шкалы MMSE, MоCА, теста рисования часов). Требуется для оценки наличия и степени выраженности когнитивного снижения. Надъядерный паралич проявляется замедленным мышлением, быстрой истощаемостью, умеренной выраженностью интеллектуальных нарушений.
- МРТ головного мозга. Выявляет расширение III желудочка, атрофические изменения среднего мозга, базальных ганглиев, премоторных зон лобной коры и височных областей. Позволяет исключить внутримозговую опухоль, энцефалит, рассеянный склероз, инсульт.
- Оценку церебральной гемодинамики. Данные о кровоснабжении мозга могут быть получены путём дуплексного сканирования, УЗДГ, МРТ сосудов. Необходимы для исключения дисциркуляторной энцефалопатии, сосудистого паркинсонизма, сосудистой деменции.
Дифференциальная диагностика осуществляется с болезнью Паркинсона, вторичным паркинсонизмом травматической, инфекционной, токсической, сосудистой этиологии, деменциями альцгеймеровского типа, поздней формой нейроакантоцитоза. От классической болезни Паркинсона надъядерный паралич отличается симметричностью паркинсонизма с момента его появления, быстрым развитием когнитивных расстройств, офтальмоплегией, ретроколлисом, выраженной атаксией, малым эффектом дофаминергической терапии. Достоверно дифференцировать прогрессирующий надъядерный паралич от прочих таупатий можно по особенностям патоморфологических изменений.
Лечение ПНП
Эффективная терапия, способная остановить прогрессирующий дегенеративный процесс, пока не найдена. Осуществляется симптоматическое лечение, направленное на облегчение состояния пациента. Проведенные фармакотерапевтические исследования не сопровождались плацебо-контролем, слабо доказывают эффективность медикаментозной терапии. В лечении когнитивных нарушений возможно применение мемантина, ингибиторов ацетилхолинэстеразы, для коррекции психоэмоциональной сферы — антидепрессантов с психоактивирующим действием (флуоксетина, пароксетина).
Большинство неврологов считают необходимым назначение стартовой дофаминергической терапии. У половины больных наблюдается определённое облегчение состояния на фоне приёма препаратов леводопы, однако данный эффект длится не более двух лет. Противопаркинсонические фармпрепараты прочих групп (ингибиторов МАО, агонистов дофаминовых рецепторов, ингибиторов КОМТ) не показали своей эффективности.
Прогноз и профилактика
При надъядерном параличе наблюдается безостановочное прогрессирование симптоматики. Проводимая терапия не оказывает существенного эффекта на течение болезни. Продолжительность жизни пациентов колеблется в пределах 5-15 лет. Летальный исход обусловлен интеркуррентными инфекциями, затяжным апноэ сна, аспирационной пневмонией. В связи с отсутствием ясного понимания этиологии и патогенеза нозологии разработка профилактических мероприятий не представляется возможной, исследования заболевания и методов его лечения продолжаются.
1. Клинические особенности надъядерного паралича/ Валикова Т.А., Алифирова В.М., Пугаченко Н.В., Цыренжапова Р.Б., Бичик А.Б.// Бюллетень сибирской медицины - 2009. - №3 (2).
2. Cлучай прогрессирующего надъядерного паралича с кортикобазальным синдромом/ Федотова Е.Ю., Чечеткин А.О., Иванова-Смоленская И.А., Иллариошкин С.Н.// Атмосфера. Нервные болезни. - 2009 - №2.
3. Трудности диагностики прогрессирующего надъядерного паралича/ Ситкали И.В., Раздорская В.В.// Бюллетень медицинских интернет-конференций. - 2015 - Т.5, №4.
4. Трудности дифференциальной диагностики прогрессирующего надъядерного паралича и болезни Паркинсона/ Магжанов Р.В., Давлетова А.И., Ибатуллин Р.А., Туник В.Ф., Идрисова Р.Ф., Бахтиярова К.З.// Анналы клинической и экспериментальной неврологии. - 2016.
Болезнь Стилла причины, симптомы, методы лечения и профилактики
Болезнь Стилла или ювенильный ревматоидный артрит — системное заболевание с воспалительным поражением суставов. Патологию диагностируют только у детей и подростков в возрасте до 16 лет, взрослым такой диагноз не ставят. О болезни Стилла говорят, когда у пациента дольше полутора месяцев протекают артриты и не удается выявить другие суставные патологии. Ювенильный артрит развивается по неустановленным причинам и дает о себе знать суставным синдромом на фоне лихорадки и высыпаний на коже. Чтобы подтвердить диагноз и пройти соответствующее лечение, нужно обратиться к ревматологу или ортопеду.
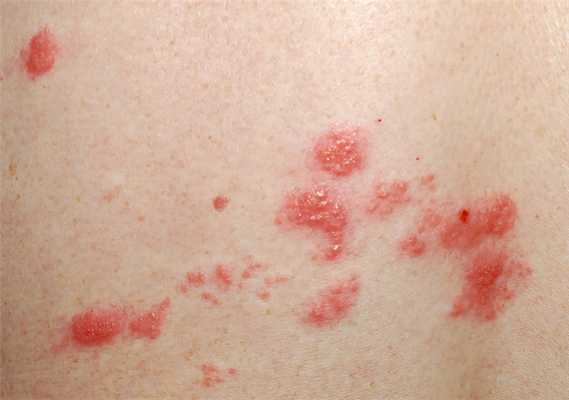
- Лихорадка, при которой температура поднимается до 39°С и выше на короткое время, а затем спадает — чаще это происходит раз в сутки, в вечернее время;
- периодические высыпания в виде розовых пятен или папул, которые локализуются на туловище, в проксимальных отделах конечностей и проявляются в периоды подъема температуры;
- суставный синдром, который вначале затрагивает один сустав и по мере прогрессирования болезни переходит в полиартрит с вовлечением крупных, мелких суставов и межфаланговых дистальных суставов кисти;
- увеличение шейных лимфоузлов, которое наблюдается у половины пациентов;
- боль в горле в виде постоянного выраженного жжения, которая характерна для начальных стадий заболевания;
- сердечно-легочные проявления, чаще всего в виде перикардита или плеврита, в редких случаях — миокардита, тампонады сердца.
Статью проверил
Дата публикации: 24 Марта 2021 года
Дата проверки: 24 Марта 2021 года
Дата обновления: 24 Октября 2022 года
Содержание статьи
Причины
Врачи не могут достоверно установить причины развития болезни Стилла. Из-за внезапного начала обострения с высокой температурой и лейкоцитозом возникло предположение об инфекционной природе заболевания. Подтвердить эту гипотезу не удается, так как единый возбудитель пока не установлен: у некоторых больных выявляют вирус краснухи, у других — цитомегаловирус. Реже обнаруживают других возбудителей — микоплазму, вирусы парагриппа и Эпштейна-Барра.
Некоторые врачи предполагают, что заболевание передается по наследству. Также существует теория о иммунологическом происхождении заболевания, согласно которой болезнь Стилла относится к аутоиммунным. Подтвердить предположение удается только в тех случаях, когда у пациентов выявляют ЦИК, которые провоцируют развитие аллергического васкулита.
Разновидности
В зависимости от характера течения и сопутствующих поражений внутренних органов, выделяют следующие формы болезни Стилла:
Системный ревматоидный артрит
Протекает в виде артрита и сопровождается симптомами поражения внутренних органов — высокой температурой в утреннее время, сыпью на лице, бедрах и в области суставов без зуда. При тяжёлом течении приводит к скоплению серозной жидкости в лёгких, сердце и других внутренних органах и, как следствие, развитию их недостаточности.
Олигоартрит
Разделяется на персистирующий, при котором за весь период развития патологии происходит поражение не более четырех суставов, и прогрессирующий. Поражает детей в возрасте от 1 года. Чем раньше появляются первые признаки заболевания, тем тяжёлее оказываются последствия. При раннем развитии болезнь провоцирует задержку роста, катаракту и слепоту, разную длину конечностей и инвалидность.
Полиартрит
Сопровождается поражением многочисленных крупных и мелких суставов по всему телу. Протекает в двух формах:
- серопозитивной — с поражением суставов рук и ног, быстрым развитием необратимых изменений;
- серонегативной — с поражением височно-нижнечелюстного сустава и суставов шейного отдела позвоночника, ранним развитием в возрасте от 1 года.
Методы диагностики
Из-за отсутствия специфических диагностических признаков выявить болезнь Стилла затруднительно. Врачу-ревматологу нужно длительное время наблюдать за пациентом, чтобы исключить другие заболевания со схожей симптоматикой. Основную диагностику с клиническими и биохимическими анализами крови, инструментальными обследованиями проводит ревматолог, который при необходимости привлекает кардиолога, пульмонолога, онколога и других профильных специалистов.
Чтобы подтвердить диагноз болезнь Стилла, в клинике ЦМРТ проводят комплексную диагностику со следующими обследованиями:
Синдром подключичного обкрадывания ( Стил-синдром )
Синдром подключичного обкрадывания — это стено-окклюзионное поражение подключичной артерии, сопровождающееся гемодинамическим реверсом в ипсилатеральной позвоночной артерии. В клинической картине характерно сочетание признаков нарушения вертебробазилярного кровотока с периодической ишемией соответствующей руки. Диагностика основана на выявлении разницы артериального давления на руках, данных ультразвуковых и рентгенологических сосудистых исследований. Малосимптомные формы заболевания являются показанием к консервативной терапии. Вариантами хирургического лечения являются шунтирование, транспозиция, ангиопластика, стентирование подключичного сегмента.


Синдром подключичного обкрадывания (стил-синдром) является одной из причин недостаточности кровообращения в вертебробазилярной системе. Впервые феномен исследован при помощи ангиографии в 1960 г. Год спустя приведено четкое развернутое описание патологии, высказано предположение о ее связи с преходящей ишемией головного мозга. В большинстве литературных источников сообщается о распространенности заболевания в пределах от 0,6% до 6,4%. Синдром в 4 раза чаще имеет левостороннюю локализацию. Соотношение мужчин и женщин среди заболевших составляет 2:1. Чаще страдают лица старше 55 лет из-за повышенной частоты атеросклероза в этой возрастной группе.
Доминирующим этиологическим триггером заболевания является атеросклеротическое поражение подключичного сегмента. Редкие причины синдрома обкрадывания включают артериит Такаясу, расслоение аорты, внешнее сдавление. Последнее возможно вследствие синдрома передней лестничной мышцы, наличия добавочного шейного ребра. Среди этиологических факторов встречаются анатомические сосудистые аномалии, деформации сосудистой стенки, в ряде случаев обусловленные дегенеративными изменениями. Описаны редкие варианты врожденного синдрома обкрадывания.
Факторы риска аналогичны таковым при атеросклерозе и включают:
- гиперлипидемию;
- артериальную гипертензию;
- сахарный диабет;
- пожилой возраст;
- курение;
- семейный анамнез сердечно-сосудистых заболеваний.
Синдром подключичного обкрадывания часто включает выраженный стеноз и/или окклюзию подключичной артерии, что приводит к снижению давления в ней дистальнее очага поражения. Более частое левостороннее поражение объясняется тем, что острый угол отхождения левой подключичной артерии увеличивает турбулентность кровотока и ускоряет развитие атеросклероза в месте соединения с аортой.
Кровоснабжаемая пораженной артерией рука получает меньший приток крови. Если дефицит кровоснабжения усугубляется физической нагрузкой, для поддержания адекватной циркуляции кровь начинает поступать из ипсилатеральной позвоночной артерии. Результатом является реверсирование кровотока в позвоночном сегменте на стороне поражения. Кровь оттекает от расположенного в задней черепной ямке базилярного сосуда, кровоснабжающего ствол, височно-затылочные отделы мозга, мозжечок. Указанные церебральные области подвергаются ишемии, что клинически проявляется вертебробазилярными симптомами.
Классификация
В современной неврологии отдельные клиницисты выделяют феномен подключичного обкрадывания как патологию, не приводящую к клиническим проявлениям, и сопровождающийся типичной клиникой синдром. Общепринята классификация патологии по тяжести возникающих гемодинамических нарушений:
- I степень (преподключичное обкрадывание) — снижение антеградного кровотока позвоночной артерии;
- II степень (перемежающаяся/частичная/латентная) — переменный кровоток: антеградный в диастолическую фазу, ретроградный в систолу;
- III степень (постоянная/расширенная) — постоянный ретроградный кровоток.
Симптомы
Клинически синдром подключичного обкрадывания проявляется признаками расстройства церебрального кровоснабжения в вертебробазилярном бассейне, сочетающимися с преходящими симптомами ишемии верхней конечности. Преподключичное обкрадывание протекает бессимптомно.
Вертебробазилярная симптоматика включает вестибулярные расстройства, признаки стволовой дисфункции, зрительные нарушения. Пациенты отмечают пароксизмальное головокружение системного типа, дискоординацию движений, неустойчивость, шаткость походки. Возможны синкопальные приступы. Стволовые нарушения характеризуются затрудненным глотанием, дизартрией, слабостью лицевой мускулатуры.
Нарушения зрения могут включать приводящий к диплопии паралич глазодвигательного нерва, дефицит зрительных полей, ухудшение остроты зрения, фотопсии. Ишемия руки часто проявляется похолоданием, онемением, парестезиями. Данные симптомы провоцируются энергичными движениями рукой, внезапным резким поворотом головы в направлении пораженной стороны.
Больные жалуются на мышечные боли и утомляемость руки при физических нагрузках. Значительная ишемия встречается редко, даже у пациентов с полной окклюзией. Предположительно, она развивается у больных с дополнительной сосудистой патологией интра- или экстракраниальных сосудов.
На фоне коронарной реваскуляризации после аорто-коронарного шунтирования подключичный стеноз влечет реверсирование кровотока из коронарного сосуда в подключичное кровообращение через протез. Развивается ишемия миокарда, клинически характеризующаяся рефрактерной нестабильной стенокардией. У таких пациентов синдром подключичного обкрадывания манифестирует приступами болей в сердце.
Физические упражнения, провоцирующие усиленный отток из церебральных артерий, вызывают транзиторную ишемическую атаку (ТИА). Назначенное неврологом своевременное лечение позволяет полностью восстановить временно утраченные нервные функции, однако повторные ТИА влекут стойкие морфологические изменения церебральных структур с развитием непреходящей симптоматики. Отсутствие адекватной терапии приводит к переходу ТИА в ишемический инсульт с развитием стойкого неврологического дефицита, инвалидизирующего больного.
Среди физикальных обследований ключевое значение имеет сравнительная тонометрия верхних конечностей, выявляющая превышающую 15 мм рт. ст. разницу давления. Истончение кожи, изменение ногтевых пластин свидетельствуют об артериальной недостаточности конечности. В неврологическом статусе типичны горизонтальный нистагм, атаксия, возможны дисфагия, расстройства речи, фациальный парез, гипестезия вовлеченной руки.
Лабораторная диагностика выявляет свидетельствующие об атеросклерозе изменения липидограммы: холестеринемию, гиперлипидемию. Среди инструментальных диагностических методов определяющее значение имеют:
- Аудиометрия. Определяется двусторонняя, практически симметричная нейросенсорная тугоухость в диапазоне высоких частот на фоне отсутствия жалоб пациента на снижение слуха.
- Стволовые акустические вызванные потенциалы. Диагностируется расстройство проводимости на медулло-понтинном уровне, свидетельствующее в пользу поражения мозгового ствола.
- Ультразвуковая допплерография (УЗДГ). Считается стандартным скрининг-методом диагностики синдрома подключичного обкрадывания. УЗГД головы и шеи определяет ретроградные изменения гемодинамики, позволяет оценить коллатеральный кровоток, извилистость сосудов, утолщение их стенок. Транскраниальная допплерография выявляет снижение гемодинамики в задних мозговых отделах, двунаправленный кровоток в базилярном сосуде.
- Дуплексное сканирование. Наряду с получением гемодинамических данных, визуализирует просвет сосуда, дает возможность диагностировать характер и степень выраженности патологических изменений. Использование манжетной пробы помогает выявить феномен скрытого обкрадывания.
- Рентгеноконтрастная ангиография. Позволяет уточнить расположение и морфологический характер сосудистого поражения. Ангиография дуги аорты считается подтверждающим синдром обкрадывания исследованием, поскольку данные ультразвуковых методов в некоторых случаях могут отображать изменения, обусловленные патологией позвоночных сосудов.
- МРТ головного мозга. Проводится для исключения прочих патологических процессов церебральной локализации, например, опухолей, аневризм, инфекционных очагов. Возможно диагностирование мелких стволовых очагов ишемии.
Дифференциальная диагностика
Церебральные ишемические симптомы стил-синдрома требуют дифференцировки с ортостатической гипотонией, синдромом позвоночной артерии, вестибулярным нейронитом, болезнью Меньера. Преходящую ишемию руки при синдроме обкрадывания следует отличать от подобных изменений, обусловленных артериитом Такаясу, фиброзно-мышечной дисплазией, болезнью Рейно, спортивной травмой. Важное значение отводится дифференциации возможных причин возникновения синдрома обкрадывания, среди которых атеросклероз, тромбоз, сдавление, сосудистые аномалии.
Лечение синдрома подключичного обкрадывания
Консервативная терапия
Рекомендована пациентам с изолированными симптомами, латентной формой заболевания. Показано амбулаторное наблюдение, периодическое УЗИ с целью контроля выраженности гемодинамических расстройств. Медикаментозное лечение синдрома направлено на компенсацию гипертонии, гиперхолестеринемии, диабета, прекращение курения, профилактику тромбообразования. Прием антитромботических средств продолжается в предоперационном периоде, а также не менее месяца после хирургического лечения.
Хирургическое лечение
Широко показано пациентам, страдающим постоянной формой синдрома с ТИА в анамнезе, с целью церебральной реваскуляризации. Выбор лечебной тактики определяется вариантом артериальной обструкции. Хирургические методы подразделяются на 2 группы:
- Открытые хирургические операции. Показаны при выраженной окклюзии, сдавлении и деформации подключичной артерии. Позволяют добиться высокой сосудистой проходимости, в большинстве случаев исключающей необходимость повторного вмешательства в будущем. Показатели проходимости достигают 95% в течение 10 лет. Наиболее популярными вариантами являются: каротидно-подключичное шунтирование, перекрестное подключично-подключичное шунтирование и транспозиция подключичной артерии.
- Эндоваскулярные вмешательства. Современные, менее травматичные операции, позволяющие минимизировать осложнения. Требуют только местной анестезии. Считаются более предпочтительными в лечении, хотя дают меньшую проходимость сосудов, чем хирургическая коррекция. К эндоваскулярным техникам относятся:
- чрескожная транслюминальная ангиопластика — является первым методом эндоваскулярного лечения, предложенным для подключичного обкрадывания. Частота рестеноза в течение 3 лет достигает 6%.
- эндоваскулярное стентирование — более современный и предпочтительный метод. Тромбоз стента встречается редко. Пятилетний показатель проходимости составляет 83-89%. Однако стентирование невозможно при полной облитерации сосуда, существенной деформации его стенки.
Бессимптомный ретроградный кровоток имеет доброкачественное естественное течение и не требуют специального лечения. Уровень смертности после хирургической реваскуляризации составляет 0,5%, частота инсульта не превышает 3,8%. Встречаемость инсульта после стентирования — до 1%, у 4,5-5% прооперированных наблюдаются незначительные осложнения.
Поскольку 95% синдрома подключичного обкрадывания обусловлены атеросклеротическими изменениями, то профилактика сводится к методам предупреждения атеросклероза. Необходимы соблюдение гипосолевой диеты с низким содержанием животных жиров, нормализация веса, отказ от курения, регулярная умеренная физическая активность.
1. Деформация подключичной артерии как причина формирования синдрома позвоночно-подключичного обкрадывания/ Кирсанов Р.И. и др.// Ангиология и сосудистая хирургия. - 2015. - 21 (2).
2. Открытые операции или эндоваскулярные вмешательства при поражениях первого сегмента подключичной артерии?/ Гавриленко А.В., Иванов В.А., Куклин А.В., Аль-Юсеф Н.Н.// Ангиология и сосудистая хирургия. - 2015. - 21(1).
3. Хирургическое лечение синдрома позвоночно-подключичного обкрадывания/ Щипакин В.Л., Процкий С.В.// Атмосфера. Нервные болезни. - 2006. - 2.
Боковой амиотрофический склероз причины, симптомы, методы лечения и профилактики
Боковой амиотрофический склероз или БАС — нейродегенеративное заболевание, поражение двигательного нейрона, которое охватывает нейроны головного и спинного мозга. Заболевание также известно как болезнь Лу Герига, Шарко, болезнь нейронов или мотонейронная болезнь.
При патологии клетки нервной системы гибнут. Если проблема охватывает нейроны спинного мозга, у пациента развивается мышечная слабость, атрофия, непроизвольные подергивания. При гибели нейронов головного мозга мышечный тонус повышается, развивается скованность мышц.
Достоверно причины заболевания неизвестны. Врачи предполагают, что оно обусловлено генетически: пациент наследует от предков ген с определенным сбоем. Пока подтвердить правильность этой гипотезы не удается, БАС считается идиопатической болезнью.

Симптомы бокового амиотрофического склероза
На начальном этапе болезнь дает о себе знать подкожными мышечными сокращениями, которые сначала локализуются в определенной группе мышц, а в дальнейшем распространяются по всему телу:
- из-за снижения количества сигналов от двигательных нейронов к мышцам последние используются меньше, постепенно теряют мышечную силу и массу
- из-за ухудшения нервной проводимости развивается мышечное напряжение, появляются спазмы
- из-за снижения физической функциональности мышц организм вынужден тратить больше энергии на поддержание их активности, что приводит к повышенной утомляемости и слабости, одышке, проблемам с дыханием
- когда пораженными оказываются мышцы лица, гортани и ротовой полости, пациенту становится сложно глотать, во рту накапливается чрезмерное количество слюны и развивается неконтролируемое слюнотечение
- на поздних стадиях болезни происходит атрофия дыхательных мышц, пациенту становится сложно дышать самостоятельно, нуждается в ИВЛ — искусственной вентиляции лёгких
- у некоторых больных случаются приступы непроизвольного смеха или плача, сильно выражены негативные эмоции — страх, печаль, беспокойство, гнев
Кроме перечисленных признаков, при БАС пациента беспокоит дискомфорт и болевой синдром. Болезненные ощущения не напрямую связаны с патологией, но развиваются из-за спазмов и напряжения мышц, сдавливания кожи.
Предположительно, патология передается генетически — люди наследуют от предков определенный ген. Подтвердить эту гипотезу данными статистики пока не удается, так как у части пациентов нет случаев заболевания в семейном анамнезе. Тогда врачи говорят о спорадической, то есть ненаследственной форме болезни моторных нейронов. К предрасполагающим факторам ее развития относятся:
- перенесённые механические травмы и шоковые состояния
- чрезмерные спортивные нагрузки, например, у профессиональных спортсменов
- длительный контакт с сельскохозяйственными химикатами, тяжелыми металлами
Достоверно известно, что заболевание не заразно, чаще всего поражает людей в возрасте 50-70 лет и чаще встречается у мужчин, чем у женщин.
Врачи называют БАС одной из разновидностей болезни двигательного нейрона. У патологии существует еще три формы, которые похожи между собой и разделяются только условно:
Прогрессирующий бульбарный паралич — разновидность БАС, которую диагностируют у четверти пациентов. Затрагивает нейроны головного и спинного мозга, поражает чаще всего женщин пожилого возраста. На ранних стадиях проявляется невнятной речью и частыми поперхиваниями при глотании. Сокращает продолжительность жизни до 6-18 месяцев с момента появления первых признаков.
Прогрессирующая мышечная атрофия поражает только нижние двигательные нейроны, провоцирует мышечную слабость и атрофию. Поражает преимущественно мужчин молодого возраста. Проявляется судорогами, слабостью и подергиванием в руках. Сокращает жизнь до 5-10 лет после появления первых симптомов.
Первичный латеральный склероз — распространяется в основном на верхние двигательные нейроны, провоцирует спастичность мышц и неустойчивость при ходьбе. Основная часть больных — мужчины старше 50 лет. Не сокращает длительность жизни, если не переходит в боковой амиотрофический склероз.
Боковой амиотрофический склероз плохо поддается диагностике из-за редкости и разнообразных симптомов, которые могут существенно отличаться у пациентов. Чаще всего пациентам рекомендуют сдать анализ крови, пройти электронейромиографию, магнитно-резонансную томографию, транскраниальную магнитную стимуляцию.
МРТ (магнитно-резонансная томография)
УЗИ (ультразвуковое исследование)
Дуплексное сканирование
Компьютерная топография позвоночника Diers
Чек-ап (комплексное обследование организма)
КТ (компьютерная томография)
К какому врачу обратиться
При первом подозрении на боковой амиотрофический склероз обратитесь к терапевту. Он соберет и проанализирует симптомы и выпишет направление к неврологу. В ходе лечения могут назначить консультацию у других врачей, например, психолога.
Лечение бокового амиотрофического склероза
БАС относится к неизлечимым болезням, вылечить его невозможно. Пациенту подбирают такой курс лечения, который замедлит гибель клеток и продлит период самостоятельного обслуживания, а также снизит выраженность симптомов.
Патогенетическое лечение
Направлено на продление жизни пациента. Сегодня врачи применяют один препарат, который замедляет прогрессирование заболевания — рилузол. Благодаря медикаменту удается продлить жизнь больного в среднем на три месяца. Применение антибиотиков, антиоксидантов, блокаторов кальциевых каналов и многих других препаратов не доказало свою эффективность при БАС.
Паллиативная терапия
Направлена на коррекцию симптомов заболевания и позволяет в некоторой степени улучшить жизнь пациента. Лечение проводят, чтобы предотвратить или на некоторое время отсрочить:
- нарушение двигательной функции
- тромбоз глубокин вен ног
- парезы и паралич
- нарушения метаболизма и атрофию мышц
- депрессию, эмоциональную нестабильность
- непроизвольное слюнотечение
- дыхательные нарушения
Болезнь всегда приводит к летальному исходу, поэтому ее осложнения условны. Осложнениями считают ухудшение состояния пациента по мере прогрессирования БАС:
- невозможность самостоятельно передвигаться, дышать и принимать пищу
- контрактуры конечностей
- болезненные мышечные спазмы
- сильное физическое истощение
- поперхивание при глотании
- депрессию
Профилактика бокового амиотрофического склероза
Поскольку достоверно причины развития БАС не установлены, врачи не могут составить эффективную специфическую программу профилактики. В целом пациентам рекомендуют вести здоровый образ жизни:
Читайте также: