Гипоплазия и аплазия тимуса. Синдром Ди Джорджи
Добавил пользователь Дмитрий К. Обновлено: 22.01.2026
Синдром Ди Джорджи - врожденный дефект, который приводит к гипоплазии или отсутствию тимуса (вилочковой железы) в сочетании с пороками развития крупных сосудов, сердца, паращитовидных желез, костей лицевого черепа и верхних конечностей
СИМПТОМЫ
В связи с тем, что так называемый «полный» синдром Ди Джорджи с серьезными нарушениями иммунитета встречается крайне редко, пациенты с данной патологией обращаются в первую очередь к другим специалистам - кардиологам и хирургам. Характерными проявлениями синдрома Ди Джорджи являются:
Пороки сердца и крупных сосудов.
Гипоплазия (недоразвитие) тимуса ведет к повышенной чувствительности у этих детей к инфекционным заболеваниям (в первую очередь вирусной и грибковой природы).
Гипоплазия паращитовидных желез проявляется развитием судорог.
Синдром также может сопровождаться другими пороками развития:
Нарушения строения лицевого черепа (у детей описывают расщелины верхней губы и нёба, широкую переносицу, высокое нёбо, широкое расстояние между бровями, большой рот).
Нарушения строения глазного яблока.
Пороки развития желудочно-кишечного тракта.
Аномалии скелета (отсутствие ногтей, дополнительные пальчики на конечностях).
Пороки центральной нервной системы проявляются задержкой психомоторного развития.
Нарушение строения гортани, трахеи.
У пациентов с этим синдромом часто встречается патология аутоиммунного характера (аутоиммунный тиреоидит - поражение щитовидной железы, цитопении - снижение количества эритроцитов, нейтрофилов, тромбоцитов), а также повышен риск развития онкологических заболеваний.
ПРИЧИНЫ
Дефект возникает в результате микрополомок на 22 хромосоме приблизительно на 8 неделе беременности. В эти сроки при внутриутробном развитии человека формируются так называемые «глоточные» щели. В результате нарушения формирования 3 и 4-й глоточных щелей (карманов) нарушается и структура органов, происходящих из них - тимуса, дуги аорты, лицевой череп, паращитовидные железы.
В норме вилочковая железа является местом, где проходят созревание Т-клетки, при отсутствии тимуса соответственно страдает Т-клеточное звено иммунной системы. В результате недоразвития паращитовидных желез нарушается регуляция электролитного баланса: прежде всего развивается снижение уровня кальция в крови.
ДИАГНОСТИКА
Общий осмотр пациента : характерные пороки развития
Общий анализ крови: снижение уровня лимфоцитов
Исследование субпопуляций лифоцитов(иммунофенотипирование): снижение количества Т-лимфоцитов
Оценка функциональной активности Т-лимфоцитов: снижение пролиферативной активности Т-лимфоцитов (способности размножаться в ответ на стимуляцию специальным агентом)
Исследование гуморального звена иммунитета: уровень иммуноглобулинов А,М,G, Е - нормальный или понижен
Рентгенологическое исследование органов грудной клетки: отсутствие тени вилочковой железы
ЛЕЧЕНИЕ
Если синдром сопровождается выраженными иммунологическими нарушениями, то для жизни таких больных, как и пациентов с тяжелым комбинированным иммунодефицитом (ТКИН), необходимы стерильные условия, назначение профилактической антимикробной и противовирусной терапии. При снижении уровня гуморального звена иммунитета проводится заместительная терапия внутривенным иммуноглобулином.
При данном заболевании эффективна пересадка эпителиальной ткани тимуса, что ведет к восстановлению количества и функций Т-лимфоцитов. Для коррекции других пороков развития также требуется хирургическое вмешательство.
КОГДА ОБРАТИТЬСЯ К ВРАЧУ
При врожденных пороках развития сердца, лицевого скелета, ЖКТ
При судорогах
При тяжелом течении вирусных инфекций (генерализованная ЦМВ-инфекция, аденовирусная, ротавирусная инфекции)
При упорном грибковом поражении слизистых оболочек
При частых отитах, повторных пневмониях.
Другие статьи
Колонка эксперта - Беллы Брагвадзе. Удивительный мир иммунитета.
Чем настоящий иммунодефицит отличается от частой простуды
Почему дети должны болеть, о бессмысленности иммуномодуляторов и о том, чем настоящий иммунодефицит отличается от частой простуды, — иммунолог Анна Щербина Иммунодефицит — это состояние, сопровождающееся значительными и долговременными изменениями в иммунной системе и серьезными симптомами. Есть иммунодефициты вторичные, а есть первичные (ПИД). Первичные обусловлены генетически. Как правило, симптомы возникают в раннем возрасте, однако иногда могут возникнуть и у взрослых. Но в любом случае проявления будут очень тяжелыми. С первичными иммунодефицитами встречаются крайне редко. Подтвердить многие такие заболевания можно, обнаружив дефект гена. Но пока, правда, найдены мутации не при всех ПИД, поиск продолжается. Текст: Дарья Саркисян Фотографии: Максим Шер Журнал "Большой город"
ЧТО ТАКОЕ ПЕРВИЧНЫЙ ИММУНОДЕФИЦИТ
Как рассказать детям об иммунитете
Презентация для школьников о том, что такое иммунитет, какие нарушения встречаются, как живут дети с первичным иммунодефицитом и как им можно помочь.
Первичный иммунодефицит. Х-сцепленный лимфопролиферативный синдром
Х-сцепленный лимфопролиферативный синдром - первичный иммунодефицит, при котором у пациентов мужского пола отмечается нарушение иммунного ответа на вирус Эпштейн-Барра.
Первичный иммунодефицит. Аутоиммунный лимфопролиферативный синдром
Аутоиммунный лимфопролиферативный синдром - первичный иммунодефицит, при котором отмечается хроническое незлокачественное увеличение лимфоузлов, печени и селезенки, аутоиммунная патология, повышение уровня иммуноглобулинов в крови.
Оптимизация диагностики и терапии наследственного ангионевротического отека у взрослых.
Особенности редкой формы первичного иммунодефицита, клинические проявления, иммунологические нарушения и принципы терапии наследственного ангионевротического отека. Индивидуальные планы самоконтроля для каждого пациента и оценкаа их эффективность. Караулов А.В., Сидоренко И.В., Капустина А.С. Первый Московский государственный медицинский университет им. И. М. Сеченова, Москва
Наследственный ангионевротический отек
Наследственный ангионевротичекий отёк - редкое, жизнеугрожающее заболевание, которое относится к группе первичных иммунодефицитов. Причина - недостаточность общего уровня или снижение функциональной активности С1-ингибитора системы комплемента. Жизнь таких больных становится кошмаром: они никогда не знают, где и когда начнется отек. Пациенты нередко испытывают страх очередного приступа, для них характерны чувство одиночества, ощущение безысходности и бесконечные проблемы на работе, в учебе и быту.
Первичный иммунодефицит. Хроническая гранулематозная болезнь
Хроническая гранулематозная болезнь (ХГБ) - генетическое заболевание, связанное с дефектом фагоцитов, клеток иммунной системы, которые защищают организм путем поглощения (фагоцитоза) вредных чужеродных частиц, бактерий, а также мертвых или погибающих клеток, из-за которого снижается их антимикробная активность.
Иммунодефицитные состояния
Слово «иммунитет» давно вошло в обиход не только врачей, но и не связанных с медициной людей: нам часто приходится слышать его в различных рекламных роликах, передачах по охране окружающей среды и т.д. Действительно, динамические изменения показателей иммунной системы происходят в ответ на различные состояния организма: инфекции, травмы, опухоли. Является ли это патологией? В большинстве случаев - нет, это нормальная реакция живого организма. Ведь основная функция иммунной системы - это борьба со всем чужеродным, будь то бактерия или опухолевая клетка. Однако в некоторых случаях изменения иммунной системы носят долгосрочный, патологический характер, такие состояния называются иммунодефицитными (ИДС).
Первичный иммунодефицит. ОВИН - общая вариабельная иммунная недостаточность
Общая вариабельная иммунная недостаточность - нарушение, характеризующееся низкими уровнями иммуноглобулинов (антител) в сыворотке крови и повышенной чувствительностью к инфекциям. Данная статья предназначена для пациентов и членов их семей и не должна заменять совета клинициста-иммунолога.
Первичный иммунодефицит. Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича является первичным иммунодефицитным состоянием, поражающим как Т- лимфоциты, так и В-лимфоциты. Также тяжело поражаются тромбоциты - клетки, помогающие останавливать кровотечение. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
Первичный иммунодефицит. Х-сцепленная агаммаглобулинемия
У больных с Х-сцепленной агаммаглобулинемией основным дефектом является неспособность предшественников В-лимфоцитов созревать до состояния В-лимфоцитов, а затем плазматических клеток. Поскольку у этих больных нет клеток, вырабатывающих иммуноглобулины, наступает тяжелая недостаточность иммуноглобулинов. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
Первичный иммунодефицит. ТКИН - тяжелая комбинированная иммунная недостаточность
Тяжелая комбинированная иммунная недостаточность (ТКИН) - самый тяжелый диагноз в списке первичных иммунодефицитов - является редким синдромом, обусловленным различными генетическими факторами, и сочетающим отсутствие функций Т- и В- лимфоцитов (а во многих случаях также отсутствие функции естественных киллеров или NK-лимфоцитов). Эти нарушения приводят к чрезвычайной чувствительности к тяжелым инфекциям. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
12 настораживающих признаков первичного иммунодефицита
ПИД не СПИД. Первичный иммунодефицит является врожденным нарушением в иммунной системе, имеющим генетическую природу. Показанием для направления к иммунологу является сочетание рецидивирующих вирусных и бактериальных инфекций либо наличие тяжелых, затяжных бактериальных инфекций. Данные Всемирной организации здравоохранения свидетельствуют о том, что частота ОРВИ 8 раз в год является нормальным показателем для детей дошкольного и младшего школьного возраста, посещающих детские учреждения.
Часто болеющие дети: чем они больны на самом деле?
Инфекции уха, горла, носа, а также бронхолёгочные инфекции составляют основной перечень заболеваний в детском возрасте. Данные ВОЗ свидетельствуют о том, что частота ОРВИ 8 раз в год является нормальным показателем для детей дошкольного и младшего школьного возраста, посещающих детские учреждения. Показанием для направления к иммунологу является сочетание рецидивирующих вирусных и бактериальных инфекций либо наличие тяжелых, затяжных бактериальных инфекций.
Иммунодефициты у детей.
Диагноз «иммунодефицит» становится все более популярным у врачей разных специальностей. Создается впечатление, что зачастую врачи, вместо того, чтобы четко определить диагноз и проводить лечение заболевания в соответствии с утвержденными стандартами, назначают иммунотропные средства, не представляя эффект и последствия такой терапии.
Диагностика семей с иммунодефицитом
Первичные иммунодефициты, являются наследуемыми заболеваниями, при которых родители являются носителями больного гена и передают его детям. В результате чего у ребенка развивается заболевание. В настоящее время в связи с развитием генетики и иммунологии известны многие гены, мутация в которых приводит к развитию различных форм первичных иммунодефицитов.
Синдром Ди Джорджи
Синдром Ди Джорджи - генетическое заболевание, относящееся к группе первичных иммунодефицитов и, наряду с ослаблением иммунитета, характеризующееся многочисленными пороками развития. Симптомами этого состояния являются частые бактериальные инфекции со склонностью к тяжелому течению, врожденные пороки сердца, аномалии развития лица и другие нарушения. Диагностика синдрома Ди Джорджи основывается на исследовании сердца, щитовидных и паращитовидных желез, изучении иммунологического статуса и данных молекулярно-генетических анализов. Лечение только симптоматическое, включает хирургическую коррекцию пороков сердца и аномалий лица, заместительную иммунологическую терапию, борьбу с бактериальными и грибковыми инфекциями.

Общие сведения
Синдром Ди Джорджи (гипоплазия тимуса и паращитовидных желез, велокардиофациальный синдром) - генетическое заболевание, обусловленное нарушением эмбрионального развития третьего и четвертого фарингеальных мешков. Впервые это состояние было описано в 1965 году американским педиатром Анджело Ди Джорджи, который классифицировал его как врожденную аплазию тимуса и паращитовидных желез. Дальнейшие исследования в области генетики помогли определить, что нарушения при этом заболевании выходят далеко за рамки первичного иммунодефицита. Это дало основание для появления другого названия синдрома Ди-Джорджи. С учетом наиболее часто поражаемых органов (небо, сердце, лицо) некоторые специалисты именуют данную патологию велокардиофациальным синдромом. Ряд современных исследователей разграничивают эти два состояния и считают, что «истинный» велокардиофациальный синдром не сопровождается выраженными иммунологическими нарушениями. Встречаемость синдрома Ди Джорджи составляет 1:3 000-20 000 - такое значительное расхождение данных обусловлено тем, что достоверная и четкая граница между этим заболеванием и велокардиофациальным синдромом до сих пор не установлена. Поэтому один и тот же больной, по мнению разных специалистов, может иметь либо первичный иммунодефицит, сопровождающийся сопутствующими нарушениями, либо многочисленнее пороки развития на фоне снижения иммунитета.

Причины синдрома Ди Джорджи
В большинстве случаев синдрома Ди Джорджи делеция 22-й хромосомы захватывает порядка 2-3 миллионов пар оснований. Чаще всего данный генетический дефект возникает спонтанно во время формирования мужских или женских половых клеток - то есть, носит герминативный характер. Лишь десятая часть всех случаев заболевания представляет собой семейную форму с аутосомно-доминантным характером наследования. Патогенез синдрома Ди Джорджи сводится к нарушению формирования особых эмбриональных образований - фарингеальных мешков (главным образом, 3-го и 4-го), которые являются предшественниками ряда тканей и органов. Главным образом, они отвечают за формирование неба, паращитовидных желез, тимуса, сосудов средостения и сердца, поэтому при синдроме Ди Джорджи возникают пороки развития именно этих органов.
Симптомы синдрома Ди Джорджи
Многие проявления синдрома Ди Джорджи определяются сразу после рождения ребенка, отдельные пороки развития (например, сердца) можно выявить еще раньше - на профилактических ультразвуковых исследованиях. Чаще всего первыми обнаруживаются аномалии развития лица - расщепление неба, иногда в сочетании с «заячьей губой», прогнатия нижней челюсти. Зачастую младенцы с синдромом Ди Джорджи имеют небольшой рот, маленький нос с расширенной переносицей, деформированные или недоразвитые хрящи ушных раковин. При относительно легком течении заболевания все вышеперечисленные симптомы могут быть выражены довольно слабо, даже расщепление твердого неба может возникать только в задней его части и выявляться лишь при тщательном осмотре у отоларинголога.
В первые месяцы жизни больного синдромом Ди Джорджи на первый план выступают проявления врожденных пороков сердца - это может быть как тетрада Фалло, так и отдельные нарушения: дефект межжелудочковой перегородки, незаращение артериального протока и ряд других. Они сопровождаются цианозом, сердечно-сосудистой недостаточностью и при отсутствии квалифицированной медицинской помощи (в том числе и хирургической) могут приводить к ранней смерти больных. Другим распространенным нарушением у детей с синдромом Ди Джорджи считаются судороги и тетания, обусловленная гипоплазией паращитовидных желез и последующей гипокальциемией.
Следующим важнейшим проявлением синдрома Ди Джорджи, отличающим его от других разновидностей велокардиофациального синдрома, является выраженный первичный иммунодефицит. Он развивается по причине аплазии или недоразвития тимуса и поэтому в большей степени затрагивает клеточный иммунитет. Однако из-за тесных взаимосвязей между гуморальным и клеточным отделами иммунной системы это приводит к общему ослаблению защитных сил организма. Больные с синдромом Ди Джорджи крайне чувствительны к вирусным, грибковым и бактериальным инфекциям, которые нередко принимают затяжное и тяжелое течение. Некоторые исследователи отмечают наличие умственной отсталости различной степени, иногда могут наблюдаться судороги неврологического происхождения.
Диагностика синдрома Ди Джорджи
Для определения синдрома Ди Джорджи применяют метод физикального общего осмотра, кардиологические исследования (ЭхоКГ, электрокардиограмма), УЗИ щитовидной железы и тимуса, иммунологические пробы. Вспомогательную роль играет проведение общего и биохимического анализов крови, изучение анамнеза больного, генетические исследования. При осмотре больных синдромом Ди Джорджи могут определяться характерные для заболевания нарушения - расщепление твердого неба, аномалии строения лица, патологии ЛОР-органов. В анамнезе, как правило, выявляются частые эпизоды вирусных и грибковых инфекций, принимающих тяжелое течение, судороги, обусловленные гипокальциемией, нередко обнаруживается обширное кариозное поражение зубов.
На ультразвуковых исследованиях вилочковой железы отмечается значительное уменьшение массы или даже полное отсутствие органа (агенезия). ЭхоКГ и другие кардиологические методы диагностики выявляют многочисленные пороки сердца (например, дефект межжелудочковой перегородки) и сосудов средостения. Иммунологические исследования подтверждают значительное падение уровня Т-лимфоцитов. Это же явление наблюдается в периферической крови и нередко сочетается с уменьшением концентрации белков-иммуноглобулинов. Биохимическое изучение крови свидетельствует о снижении уровня кальция и гормонов паращитовидной железы. Врач-генетик может выполнить поиск делеций в 22-й хромосоме посредством флуоресцентной гибридизации ДНК или мультиплексной полимеразной цепной реакции.
Лечение синдрома Ди Джорджи
Специфического лечения синдрома Ди Джорджи на сегодняшний момент не существует, используют только паллиативные и симптоматические методики. Очень важно как можно раньше выявить врожденные пороки сердца и при необходимости произвести их хирургическую коррекцию, поскольку именно сердечно-сосудистые нарушения являются наиболее частой причиной неонатальной смерти при этом заболевании. Значительную опасность представляют собой судорожные приступы, обусловленные гипокальциемией, что требует своевременной коррекции электролитного баланса плазмы крови. Помощь хирургов при синдроме Ди Джорджи также может потребоваться для устранения пороков развития лица и неба.
Из-за выраженного иммунодефицита любые признаки бактериальной, вирусной или грибковой инфекции являются поводом для срочного применения соответствующих препаратов (антибиотиков, противовирусных и фунгицидных средств). Для улучшения иммунного статуса больного синдромом Ди Джорджи может производиться заместительное вливание иммуноглобулинов, полученных из донорской плазмы. В отдельных случаях осуществлялась пересадка вилочковой железы, которая стимулировала образование собственных Т-лимфоцитов - это способствовало улучшению качества жизни больных.
Прогноз и профилактика синдрома Ди Джорджи
Прогноз синдрома Ди Джорджи большинством исследователей оценивается как неопределенный, так как данное заболевание характеризуется значительной вариабельностью симптомов. В тяжелых случаях имеется высокий риск ранней неонатальной смерти из-за сочетания сердечно-сосудистых и иммунологических нарушений. Более доброкачественные формы синдрома Ди Джорджи требуют достаточно интенсивной паллиативной терапии, особенно важно уделять внимание лечению и профилактике вирусных и грибковых инфекций. Интеллектуальное развитие больных несколько замедлено, однако при правильной педагогической и психологической коррекции проявления задержки развития можно нивелировать. Из-за частого спонтанного характера мутаций профилактика синдрома Ди Джорджи не разработана.
Септооптическая дисплазия
Септооптическая дисплазия - врожденное заболевание, относящееся к порокам прозэнцефалической группы, характеризуется аномалиями развития зрительного нерва, гипофиза и прозрачной перегородки. Симптомами этого состояния являются нистагм и другие зрительные нарушения, признаки эндокринных расстройств (задержка роста и полового созревания), возможно развитие умственной отсталости. Диагностика септооптической дисплазии производится на основании данных общего осмотра больного, офтальмологических исследований, компьютерной и магнитно-резонансной томографии головного мозга, а также молекулярно-генетических анализов. Специфическое лечение не разработано, симптоматические мероприятия включают в себя коррекцию зрения и заместительную гормональную терапию при эндокринных расстройствах.

Септооптическая дисплазия (синдром де Морсье) - врожденное нарушение развития головного мозга и зрительного аппарата различной (в том числе и генетической) природы. Причинами этого состояния, помимо генетических мутаций, могут выступать инфекции матери во время вынашивания ребенка, молодой возраст родителей, сосудистые нарушения у женщины. Название «септооптическая дисплазия» ввел французский педиатр Де Морсье, который в 1956 году составил наиболее обширное и полное описание этого состояния. В настоящий момент под синдромом Де Морсье врачи-генетики подразумевают только наследственную форму септооптической дисплазии. На сегодняшний день общая встречаемость этого заболевания (как приобретенных, так и наследственных форм) составляет примерно 1 случай на 10 000 новорожденных, мальчики и девочки поражаются с одинаковой частотой. Выраженность септооптической дисплазии может значительно отличаться у разных больных - от практически полного отсутствия симптомов до тяжелых нарушений, осложненных ДЦП, отставанием в физическом и умственном развитии.

Причины септооптической дисплазии
Септооптическая дисплазия наследственного характера развивается по причине мутаций в гене HESX1, расположенного на 3-й хромосоме. Этот ген принадлежит к обширному классу гомеобоксных генов, принимающих активное участие в регуляции процессов эмбриогенеза. В частности, HESX1 регулирует эмбриональное развитие структур головного мозга (прозрачной перегородки, гипофиза, хиазмы, зрительного нерва), поэтому его мутации приводят к развитию септооптической дисплазии. Кроме этого, доказано участие данного гена в регуляции осевой и двухсторонне-симметричной структуры тела. На сегодняшний день выявлено четыре типа миссенс-мутаций гена HESX1, наличие которых обуславливает появление признаков септооптической дисплазии, все они наследуются по аутосомно-рецессивному типу.
Выраженность симптомов заболевания может существенно различаться - от незначительных зрительных нарушений (миопии, косоглазия) до яркой клинической картины со слепотой, гипопитуитаризмом и тяжелой умственной отсталостью. Исследователи пока не нашли взаимосвязи между типом мутации HESX1 и тяжестью симптомов септооптической дисплазии. Возможно, развитие этого заболевания является результатом совокупного влияния как внутренних (генетических), так и внешних факторов. Также остается неясным то обстоятельство, что у детей молодых матерей (в возрасте менее 23 лет) септооптическая дисплазия возникает чаще, а ее наследственная разновидность протекает намного тяжелее.
Патогенез септооптической дисплазии заключается в нарушении процесса дифференцировки эмбриональных тканей в области зачатков гипофиза, хиазмы, мозолистого тела и других структур мозга. По этой причине данное заболевание в тяжелых случаях может сопровождаться другими неврологическими симптомами - детским церебральным параличом, умственной отсталостью. Практически всегда при септооптической дисплазии выявляются те или иные нарушения зрения и эндокринные расстройства, обусловленные пороками развития гипофиза. Из-за этого возможно возникновение вторичных патологий, вызванных аномальной функцией желез внутренней секреции.
Симптомы септооптической дисплазии
Возраст появления симптомов септооптической дисплазии сильно варьирует у разных больных, в тяжелых случаях диагноз может быть поставлен в первые дни и месяцы жизни, при стертой форме заболевания - лишь в младшем или даже старшем детском возрасте. Обычно первым проявлением патологии становится развитие горизонтального нистагма, обусловленное гипоплазией зрительного нерва. Еще раньше при осмотре ребенка могут определяться признаки эндокринной недостаточности гипофиза: гипогликемия, уменьшенный размер половых органов, аномальная желтуха. В редких случаях в первые месяцы жизни при септооптической дисплазии возникают судорожные припадки, длительное сохранение транзиторных рефлексов и другие неврологические нарушения.
По мере роста ребенка, страдающего септооптической дисплазией, может выявляться отставание как в физическом, так и в интеллектуальном развитии. Патологии зрения нарастают, нередко случаются эпилептические припадки. У таких больных обычно раньше, чем у сверстников, начинается процесс полового созревания, обусловленный эндокринными расстройствами. Однако в ряде случаев эндокринные нарушения могут быть выражены довольно слабо или совсем отсутствовать. Такая же ситуация и с задержкой психического развитием больных септооптической дисплазией - она колеблется от нормального интеллекта до глубокой умственной отсталости. Последняя может указывать на наличие сопутствующих пороков развития головного мозга - голопрозэнцефалии, гипоплазии мозолистого тела. Непостоянство симптомов и различная степень их выраженности значительно осложняет диагностику септооптической дисплазии.
Диагностика и лечение септооптической дисплазии
Для определения септооптической дисплазии используются результаты общего осмотра больного, неврологические и офтальмологические исследования, магнитно-резонансная и компьютерная томография, молекулярно-генетические анализы. При осмотре может обнаруживаться отставание в физическом развитии (у детей раннего возраста), раннее наступление полового созревания (у подростков), признаки множественной гормональной недостаточности. Офтальмологическое обследование может выявить нистагм и признаки гипоплазии зрительного нерва - уменьшение размеров диска зрительного нерва, ослабление всех пиков на ЭРГ, полную слепоту. Однако по клинической картине диагностировать септооптическую дисплазию достаточно проблематично, поскольку это состояние характеризуется значительной вариабельностью симптомов.
На магнитно-резонансной томографии определяется отсутствие или выраженное недоразвитие прозрачной перегородки, аплазия или гипоплазия зрительного нерва, в ряде случаев - недоразвитие мозолистого тела. Выявляются нарушения в формировании гипофиза, в особенно тяжелых случаях септооптической дисплазии могут обнаруживаться другие пороки центральной нервной системы, например, голопрозэнцефалия. Биохимический анализ крови позволяет подтвердить наличие недостаточности гормона роста (соматотропина) и других гормонов гипофиза. Молекулярно-генетическая диагностика осуществляется врачом-генетиком, производится прямое секвенирование гена HESX1 с целью подтверждения мутаций. Отсутствие генетических дефектов не является поводом для исключения септооптической дисплазии, так как заболевание может возникать вследствие причин ненаследственного характера. «Золотым стандартом» в диагностике этого состояния являются данные МРТ и КТ.
Специфического лечения септооптической дисплазии не существует, применяют симптоматические и паллиативные лечебные мероприятия. При наличии эндокринных нарушений назначают заместительную терапию, схема которой зависит от характера гормональной дисфункции, что определяется в рамках анализа крови. В большинстве случаев септооптической дисплазии нарушения зрения практически не поддаются коррекции. Симптоматическая терапия включает в себя применение противосудорожных препаратов (при эпилептических припадках) и ноотропных средств, работу детских психологов с больным ребенком при умственной отсталости.
Прогноз и профилактика септооптической дисплазии
Прогноз септооптической дисплазии чаще всего неопределенный из-за сильной вариабельности проявлений заболевания. При наличии тяжелой гипоплазии зрительных нервов, гипофиза, прозрачной перегородки существует крайне высокий риск летального исхода в раннем детстве из-за многочисленных нарушений. Аналогично ухудшают прогноз септооптической дисплазии сопутствующие патологии - детский церебральный паралич, голопрозэнцефалия. Во многих случаях заболевание протекает достаточно благоприятно - симптомы ограничиваются незначительными нарушениями зрения, иногда наличием слабо выраженной умственной отсталости, повышенным риском судорожных припадков. Профилактика септооптической дисплазии возможна только в отношении форм заболевания, обусловленных ненаследственными причинами - инфекционными болезнями матери, ее молодым (юным) возрастом, сосудистыми нарушениями во время беременности.
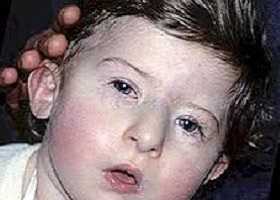
Синдром Ди Джорджи еще в некоторых источниках указывают как синдром Ди Георга. Впервые патология была описана в 1965 году американским педиатром и эндокринологом — Анджело Мария Ди Джорджи (фото). По международной классификации десятого пересмотра (МКБ-10) данному иммунодефициту был присвоен код D82.1 и указан как синдром дивертикула глотки, вилочковой железы: алимфоплазия, аплазия либо гипоплазия с иммунной недостаточностью.

Синдром Ди Джорджа как первичный иммунодефицит является редким врожденным заболеванием (1 случай на 4-6 тыс. человек) и относится к идиопатическому изолированному гипопаратиреозу - состоянию, для которого характерно снижение выработки паратгормонов и гипокальциемия. Для синдрома свойственно множество фенотипов поражения, осложнений и присоединения сопутствующих нарушений.
Патогенез
Для синдрома Ди Георга характерна аплазия или гипоплазия вилочковой железы (тимуса), а также агенезия или дисгенезия паращитовидных желез в результате нарушений закладки и эмбриональной дифференцировки 3-4 жаберных (глоточных) карманов. Это вызывает резкое снижение популяции Т-лимфоцитов, нарушение их дифференцировки и как следствие — иммунологическую недостаточность. У детей могут развиваться различные врождённые аномалии крупных сосудов, например, дефекты аорты, межжелудочковой перегородки или синий порок сердца.
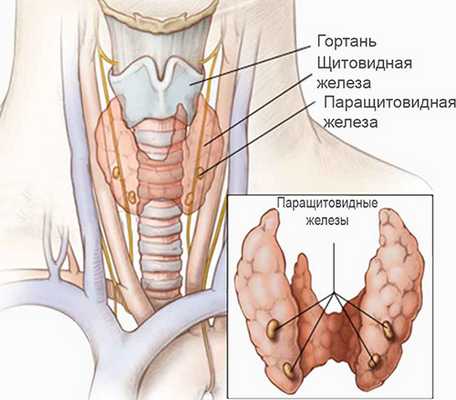
Расположение паращитовидных и щитовидных желез
Классификация
В зависимости от спектра клинических проявлений и врожденных аномалий синдром Ди Георга может быть полным и частичным, к примеру, протекать как изолированная недостаточность паращитовидных желёз либо врождённое отсутствие паращитовидных (околощитовидных) желез, которые приводят к гипокальциемическим судорогам, наблюдающимся у новорожденных в виде неонатальной тетании. Нарушения закладки тимуса приводят к развитию различных инфекционных заболеваний и обычно вызваны иммунологической недостаточностью.
Достаточно часто болезнь протекает в менее тяжелой форме и является наиболее распространенной причиной умственной отсталости с малым количеством других проявлений, поэтому может быть формально не диагностирована.
Причины
У синдрома Ди Джорджи или врождённой аплазии тимуса и паращитовидной железы генетическая причина развития. Патология развивается в результате делеции центрального участка более длинного плеча двадцать второй хромосомы, поэтому еще обозначается как синдром 22q 11.2. Однако, в некоторых случаях аналогичная клиническая картина наблюдалась и после других хромосомных перестроек - транслокаций и микроделеций, развивающихся в таких хромосомах как ТВХ1, 10р13, 17р13, 18q21 и пр. Эти мутации спорадические в 90% и обычно происходят на этапе мейоза спермато— или овогенеза.
Для данного синдрома дисэмбриогенеза 3-4 жаберной дуги — фенотипа CATCH 22 характерно наследование по аутосомно-доминантному типу, которое наблюдается лишь в 5-10% случаев, но при этом не исключается возможность аутосомно-рецессивного типа наследования с разной экспрессивностью.
Симптомы врождённой аплазии тимуса и паращитовидных желёз (Синдром Ди Джорджи)
Синдром Ди Джорджи - это первичный иммунодефицит, вызванный врожденным отсутствием или наличием аномалий развития тимуса и паращитовидных желез, который отличается триадой клинических симптомов, включающей:
- различные врожденные пороки сердца, дефекты крупных сосудов, носа, рта, ушей, что в результате дает специфические черты лица (как на фото): гипертелоризм (увеличенное расстояние между глазами), микрогнатия (недоразвитие нижней челюсти), низкое расположение ушных раковин;
- первичный иммунодефицит - нарушен как клеточный, так и гуморальный ответ иммунитета в результате аплазии вилочковой железы и невозможности обеспечения нормального развития Т-клеток;
- прогрессирующая гипокальциемия (пониженная концентрация кальция в кровотоке) - вызвана гипопаратиреозом, сопровождается судорожным синдромом и развитием скелетных аномалий.
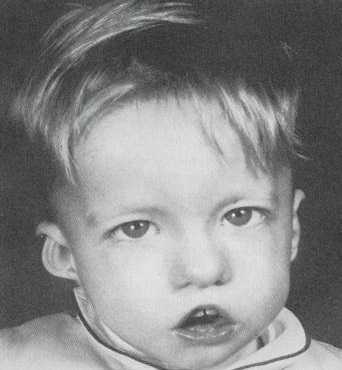
Ребенок со специфическими чертами, характерными для синдрома Ди Джорджи
Особенности синдрома могут широко варьироваться даже в пределах одной семьи и затрагивать различные системы органов. Сопутствующими проблемами становится:
- нарушение работы почек и их атрофия, развитие нефрокальциноза, гидронефроза и пр.;
- проблемы с пищеварением и перистальтикой ЖКТ;
- дефицит гормона роста;
- проблемы с речью;
- психические расстройства, в том числе шизофрения;
- потеря слуха (проводящая и нейросенсорная обычно вызвана черепно-лицевыми синдромами);
- нарушения координации, которые могут быть вызваны гипоплазией мозжечка;
- синюшность кожных покровов в результате плохого кровообращения; ;
- частые переломы костей;
- болезнь Грейвса; .
Анализы и диагностика
Для подтверждения диагноза синдром Ди Джорджа необходимо выявление агенезии или дисгенеза паращитовидных желёз, аплазии вилочковой железы, иммунологической недостаточности, черепно-лицевых дисморфий (микрогнатии, гипертелоризма, антимонголоидного разреза глаз, расщелины губы и нёба, деформированных и/или низко расположенных ушных раковин) и прочих типичных аномалий развития. При этом оказываются эффективными различные исследования, включая УЗИ, МРТ и пр., а также используются методы генетического тестирования, к примеру, FISH (метод флуоресцентной гибридизации in situ).
Наиболее яркими проявлениями является гипопаратериоз и молочница. Также при плановом обследовании выявляются дефекты аорты, тетрада Фалло, катаракта, паховая грыжа и тому подобное. При помощи серологических исследований можно выявить лимфоцитопению, гипокальциемию, гипоγ-глобулинемии. Благодаря иммунологическим методам удается обнаружить нарушения трансформации лимфоцитов и их функциональной активности, в 20% случаев наблюдается сниженное количество Т-клеток. После иммунологических исследований важно провести дифференциальный диагноз с прочими первичными иммунодефицитами, таким как: синдром Вискотта-Олдрича, Брутона.
Лечение
Так как недуг неизлечим больным обычно назначают симптоматическое и комплексное поддерживающее лечение. Основными приемами считается соблюдение диеты, стерильные условия, прием кальция, комплексов витамин, антибиотиков, противогрибковых, иммуномодулирующих, седативных препаратов и пр.
Синдром Денди-Уокера
Синдром Денди-Уокера - это врожденная патология нервной системы, для которой характерна триада признаков: гидроцефалия, гипоплазия или аплазия мозжечка, кисты задней черепной ямки. Болезнь имеет полиэтиологическую природу, среди провоцирующих факторов выделяют генетические аномалии, тератогенные влияния. Клиническая симптоматика включает классические признаки гидроцефалии, многообразные неврологические нарушения. Диагностика порока требует проведения нейросонографии, МРТ, эхокардиографии, также возможна пренатальная постановка диагноза при УЗИ-скрининге. Лечение заключается в консервативных и хирургических методах коррекции гидроцефалии.
МКБ-10


Пороки ЦНС занимают второе место в группе врожденных болезней, уступая только сердечно-сосудистым аномалиям. До 80% всех патологий сопровождаются гидроцефалией, как при синдроме Денди-Уокера. Данное заболевание было описано американским нейрохирургом В. Денди в 1914 году, спустя 28 лет канадско-американский нейрохирург А. Уокер с коллегами предложил оперативный способ лечения порока. Патология встречается с частотой 1:25000-1:35000, девочки болеют чаще. Среди младенцев с врожденной гидроцефалией патология регистрируется в 3,5-12% случаев.
Этиологические факторы синдрома Денди-Уокера до сих пор точно не выяснены. По современным данным, патология имеет мультифакториальную природу, ее развитию способствуют как внутренние причины (хромосомные и генные мутации), так и воздействие экзогенных тератогенов. Провоцирующими факторами заболевания могут выступать:
- вирусные инфекции (цитомегаловирус, краснуха);
- гестационный диабет;
- употребление алкоголя матерью во время беременности.
Новейшие исследования показывают четкую связь изолированной формы аномалии Денди-Уокера с мутациями генов ZIC1 и ZIC4. Риск развития заболевания повышается у младенцев с врожденными нарушениями обмена веществ. Чаще всего синдром ассоциирован с одной из форм 3-метилглутаконовой ацидурии, которая проявляется ретинопатией, почечной дисфункцией, метаболическим ацидозом и повышением активности печеночных ферментов.
Выделяют три основные гипотезы структурных поражений головного мозга при болезни Денди-Уокера. Согласно первой из них, пороки ЦНС вызваны замедлением эмбрионального развития на раннем этапе при закладке ромбовидного мозга. Такая ситуация наблюдается под действием тератогенных факторов либо на фоне генетических мутаций. Вторая гипотеза указывает на заращение выходного отверстия 4 желудочка мозга и позднее открытие апертуры Мажанди.
Новые экспериментальные данные подтверждают вероятность третьей гипотезы: типичные анатомические изменения развиваются при возникновении сосудистого сплетения на тонкой крыше ромбовидного мозга. Патология сопровождается внутриутробной гидроцефалией, вызывает серьезные неврологические нарушения. На фоне этого развивается большая киста в задних отделах черепа, которая препятствует формированию мозолистого тела и червя мозжечка.
Специалисты выделяют открытую и закрытую формы синдрома. В первом случае наблюдается окклюзия отверстий Люшка и Мажанди, что сопровождается серьезными нарушениями ликвородинамики. Второй вариант не имеет таких патологий, поэтому протекает в более благоприятной форме. В нейрохирургии важное значение имеет анатомическая классификация порока, согласно которой выделяют 2 формы синдрома Денди-Уокера:
- Полная. Характеризуется отсутствием червя мозжечка, наличием связи между полостью четвертого желудочка и мозжечково-мозговой цистерной.
- Неполная. Проявляется частичным недоразвитием мозжечкового червя в его нижней части, за счет чего коммуникация с большой цистерной реализуется не на всем протяжении.
В 1989 г. американским педиатром и нейрорадиологом Джеймсом Барковичем была предложена альтернативная классификация, базирующаяся на результатах МР-диагностики. Выделена группа заболеваний, названная «комплексом Денди-Уокера», который включает 4 клинических формы: классическую аномалию (полная форма), вариант Денди-Уокера (менее грубые нарушения), кисту кармана Блейка и mega cisterna magna.
Симптомы
Основное проявление синдрома Денди-Уокера - гидроцефалия, возникающая в первые месяцы жизни ребенка. Степень выраженности симптоматики зависит от варианта синдрома, скорости прогрессирования нарушений, общего состояния новорожденного. К ранним признакам относят беспокойное поведение, монотонный крик, нарушение питания и частые срыгивания. Внешне обнаруживается выбухание родничков, расширение черепных швов, быстрое увеличение окружности головы.
Характерный признак гидроцефалии при синдроме Денди-Уокера - симптом заходящего солнца: при взгляде вниз радужка частично скрывается нижним веком, над ней появляется белая полоска склеры. Неврологические симптомы также представлены нистагмом, экзофтальмом, косоглазием. Нередко у новорожденных отмечается судорожный синдром, парезы и параличи конечностей. При прогрессировании болезни характерно отставание в психомоторном развитии.
Осложнения
В 65-68% случаев синдром Денди-Уокера сопровождается другими структурными неврологическими аномалиями. Чаще всего диагностируется агенезия мозолистого тела, стеноз Сильвиева водопровода, гетеротопия извилин коры мозжечка. При тяжелых формах заболевания определяется недоразвитие ствола головного мозга, что сопряжено с глубоким угнетением витальных функций и высоким риском младенческой летальности.
До 55% детей имеют сопутствующие врожденные синдромы: Уокера-Варбурга, PHACE, Ritscher-Schinzel. Характерны различные формы хромосомных аберраций: делеции, дупликации, трисомии и триплоидии. Поражение кардиоваскулярной системы проявляется септальными дефектами и нарушениями постнатальной гемодинамики. Реже определяются офтальмологические пороки: ретинальная дисплазия, микрофтальмия.
Диагностика
Первичное обследование ребенка с неврологическими нарушениями проводится врачом-педиатром или неонатологом. При гидроцефалии и подозрении на синдром Денди-Уокера к диагностике подключается детский невролог, нейрохирург. Физикальный осмотр выявляет неспецифические признаки внутричерепной гипертензии, нарушения моторного развития, сопутствующие аномалии. Для постановки окончательного диагноза проводится:
- Нейросонография. При ультразвуковом сканировании головного мозга определяется крупное кистозное образование в задней части черепной коробки. На УЗИ мозжечковый червь не визуализируется, недоразвитые полушария мозжечка раздвинуты, желудочковая система мозга расширена и деформирована.
- МРТ головного мозга. Магнитно-резонансная томография в сагиттальной и аксиллярной проекции демонстрирует расширение четвертого желудочка, грубые нарушения развития мозжечка, другие структурные аномалии.
- Эхокардиография. УЗИ сердца рекомендовано всем детям с синдромом Денди-Уокера, поскольку он нередко ассоциирован с врожденной кардиальной патологией. По результатам ЭхоКГ можно определить аномалии развития клапанного аппарата, дефекты внутрисердечной перегородки, патологии расположения крупных сосудов.
- Кариотипирование. При комбинированных врожденных пороках развития необходима консультация генетика и тщательное изучение генетического материала ребенка. Углубленное исследование направлено на диагностику хромосомных аберраций.
- Пренатальная диагностика. При проведении УЗИ плода удается предположить аномалию на сроке 15-16 недель, более четкая визуализация структур IV желудочка возможна после 22 недели гестации. При сочетании патологии с расщелинами лица эхосонография может быть недостаточно информативна.
Дифференциальная диагностика
При постановке диагноза необходимо дифференцировать синдром с гипоплазией мозжечка другой этиологии, ретроцеребральными кистами и расширениями большой мозговой цистерны. Патогномоничным признаком синдрома Денди-Уокера считается дефект червя мозжечка, который не возникает при других вариантах пороков ЦНС. Проводится дифференциальная диагностика с арахноидальными кистами, которая требует дополнительных инструментальных методов исследования.
Лечение синдрома Денди-Уокера
Консервативная терапия
Наибольшую опасность для жизни и здоровья младенца представляет гидроцефалия, для коррекции которой показано лечение в отделении интенсивной терапии. Назначается массивная дегидратационная терапия, которая включает разные типы диуретиков, гипертонические инфузионные растворы. Усилия врачей направлены на уменьшение ликворопродукции и нормализацию внутричерепного давления, чтобы предупредить отек мозга и компрессию церебральных структур.
Хирургическое лечение
При неэффективности консервативных методов и неуклонном прогрессировании гидроцефалии требуется помощь детских нейрохирургов. Основным методом коррекции при синдроме Денди-Уокера являются шунтирующие операции с имплантацией дренажных трубок или клапанных регулируемых систем. Существует несколько типов шунтирования: вентрикуло-перитонеальное, вентрикуло-атриальное, вентрикулоцистерностомия.
Для коррекции окклюзии ликворных путей применяются методы эндоскопической вентрикулостомии, которые восстанавливают нормальный отток цереброспинальной жидкости, устраняют острую неврологическую симптоматику. При гипертензионно-гидроцефальном кризе показана вентрикулярная разгрузочная пункция, которая относится к вариантам экстренного временного дренирования.
Прогноз и профилактика
Исход заболевания зависит от глубины поражения ЦНС, скорости нарастания гидроцефалии, наличия сочетанной патологии. Нарастающая гипертензия и грубые неврологические пороки в 90% случаев становятся причиной смерти пациента в первые годы жизни, для выживших детей с неполным вариантом аномалии прогноз более благоприятный. Вследствие недостаточной изученности этиопатогенеза болезни эффективные меры профилактика пока не разработаны.
1. Случая мальформации Денди-Уокера с доношенной беременностью и родами / Е.В. Беляева, Л.В. Лапшина, Е.В. Шапошникова, А.А. Молгачев // Журнал неврологии и психиатрии. - 2018. - №2.
2. Аномалия Денди-Уокера - редкая причина сирингомиелии у взрослых /Г.Ю. Евзиков// Неврология, нейропсихиатрия, психосоматика. - 2017. - №9.
3. Синдром Денди-Уокера у новорожденных/ Л.А. Петрова, А.В. Розанов, Ю.И. Барашнев, В.О. Панов// Российский вестник перинатологии и педиатрии. - 2010. - №1.
Читайте также: