Лечение мышечной дистрофии Дюшенна и Беккера у детей. Физиотерапия
Добавил пользователь Morpheus Обновлено: 21.01.2026
Прогрессирующие мышечные дистрофии — группа заболеваний, общим симптомом которых является слабость, атрофия скелетных мышц, нарушениям функции опоры и движения, контрактурам. На 45-60% мышечные дистрофии носят наследственный характер. Реабилитация при мышечной дистрофии нераздельно идет вместе с лечением. Рассмотрим основы и основные направления.
Основной целью реабилитации детей с мышечной дистрофией является, возможно, раннее выявление и лечение, компенсация нарушенных функций опорно-двигательного аппарата, максимально возможное сохранение двигательных функций, подготовка ребёнка к жизни в обществе.
При составлении программы реабилитации основное направление — функциональное. Цель — максимально развить или хоть как-то сохранить двигательные способности ребёнка. В составлении программы участвуют врачи разных специальностей и всех необходимых профилей. Учитывают форму, стадию болезни, механизм нарушения двигательных функций.
Реабилитационные мероприятия начинаются с первых дней и, учитывая тяжесть состояния, могут проводиться всю оставшуюся жизнь. Медицинская реабилитация неразрывно связана с педагогикой и психологией, причём применительно не только к ребёнку, но и к взрослым членам семьи.
Этапы реабилитации при прогрессирующей мышечной дистрофии
- Первый этап реабилитации при ПМД начинается в стационаре, после полного обследования ребёнка. По сути, на этом этапе проводится восстановительное лечение, способствующее стабилизации процесса. Длительность его может продолжаться до 30—35 дней, в зависимости от стадии болезни, темпа и прогрессирования. На этом этапе проводится большое количество процедур, стимулирующих мышечную активность — миостимуляция, гидрокинезотерапия, парентеральное введение препаратов).
- Второй этап — санаторно-курортный, осуществляется в местном санатории или на ближнем курорте. Задачи этого этапа — поддержание и развитие достигнутого эффекта применением природных физических факторов. Это физиотерапия в сочетании с климатолечением, грязелечением, лечебной физкультурой.
- Третий этап реабилитации — амбулаторное лечение. На этом этапе все возможные мероприятия и физиопроцедуры проводятся дома. В условиях поликлиники можно провести курс физиотерапии, массажа и ЛФК 2—3 раза в год.
Задачей реабилитации является не только необходимость оказать медицинскую помощь, но также помочь обучению, стать социально независимым, получить профессию.
Дифференцированный реабилитационный комплекс при ПМД
Программный комплекс реабилитации разрабатывается в зависимости от формы, стадии болезни, возраста ребёнка. Комплекс может включать в себя медикаментозные, ортопедические, физиотерапевтические методы, массаж и лечебную физкультуру. Важное значение для больных ПМД имеет двигательный режим.Необходимо сохранять все доступные движения, так как гипокинезия приводит к прогрессированию атрофии, контрактур.
Также не менее важными являются диета, медикаментозное лечение. При подборе диеты основной задачей является необходимость возмещения дефицита белка. Содержание белка в пище увеличивают до 110 — 140 гр при норме 105 в виде творога, рыбы, отварного мяса. Медикаментозное лечение при таком заболевании проводится постоянно, практически на протяжении всей жизни. Основными препаратами при ПМД являются анаболические стероиды, назначаемые с целью снижения распада мышечных белков, а также аминокислоты, которые принимают с целью восполнения потерь организмом белка.
Лечебная физкультура
В прежние годы ЛФК при миопатиях считалась противопоказанной. Однако клинические исследования показали, что дозированная и целенаправленная физическая нагрузка , не усиливающая распада мышечной ткани, является стимулятором обмена, необходимым для правильного развития детей. Также физическая активность является профилактикой контрактур и деформаций костной ткани. Виды ЛФК и темп занятий подбирается индивидуально.
Основные виды физической нагрузки, которые назначаются больным с ПМД:
- утренняя гимнастика
- лечебная гимнастика
- прогулки и игры
- гидрокинезотерапия
Утреннюю гимнастику проводят 8—10 минут до завтрака, в медленном темпе, периодически меняя комплекс физических упражнений. Можно проводить занятия в игровой форме.
Лечебная гимнастика включает общеукрепляющие и щадящие упражнения, для тренировки поражённых мышц и сохранения объёма движения. Обязательно включают дыхательные упражнения, способствующие выработке глубокого вдоха и усиленного выдоха. Можно использовать различные вспомогательные приспособления. Продолжительность лечебной гимнастики составляет от 15 до 30 минут в зависимости от состояния ребёнка.

Гидрокинезотерапия — физические упражнения в воде и бассейне является преимущественным видом физической нагрузки для детей, страдающих мышечной дистрофией. Упражнения в тёплой воде способствуют релаксации мышц, улучшению кровоснабжения, влияют на ослабленные мышцы и способствуют восстановлению объёма движений. Водная среда облегчает статическую нагрузку, помогает сохранить вертикальную позу, укрепить опорную функцию, стимулировать ходьбу.
Занятия в бассейне проводят 2 — 3 раза в неделю. Предварительно можно провести небольшую разминку в зале. Занятие состоит из 5—7 минут свободного плавания, затем 10 минут упражнений с участием методиста, подводного массажа и свободного плавания.
Лечебное плавание в бассейне проводят при температуре воды 29 — 30 градусов. Рекомендуется детям при лёгкой степени поражения, медленном прогрессировании болезни.
Массаж при ПМД
Массаж также активно назначают и используют при различных формах мышечной дистрофии. При первичной форме назначают лёгкий, поверхностный массаж поражённых мышц и общий, с использованием приёмов поглаживания и растирания. Массаж вызывает реакцию нервно-сосудистой системы, усиливает крово- и лимфообращение, насыщение кислородом и питание тканей.
В первую очередь проводят массаж спины и конечностей (10—15 минут, до 30 процедур на курс, до 5 курсов в течение года). Дома массаж могут проводить родители, которых обучили специалисты.
Подводный ручной массаж в тёплой воде при контрактурах суставов усиливает релаксацию, кровообращение. Используют приёмы поглаживания, лёгкого разминания.
Массаж часто сочетают с другими методами физической терапии. При разработке контрактур сначала рекомендуют тепловую процедуру, потом массаж. После массажа при необходимости назначают процедуру электролечения (Электрофорез или СМТ). Очень важно квалифицированно подбирать приёмы массажа со строго индивидуальной дозировкой.
Природные физические факторы при лечении мышечной дистрофии
Физические факторы также широко назначают при прогрессируемой мышечной дистрофии. Они оказывают общеукрепляющее воздействие, улучшают обменные процессы, кровоснабжение, трофику мышц и тканей. Рассмотрим наиболее часто рекомендуемые природные физические факторы при ПМД:
- Аэротерапия и климатолечение. Основа лечебного воздействия — пребывание ребёнка на свежем воздухе. Воздушные ванны проводят в сочетании с лёгкими физическими упражнениями. Прогулки, лёгкие игры на воздухе способствуют снижению гипоксии, закаливанию.
- Профилактические ультрафиолетовые облучения — показаны в осенне-зимнее время. Облучения проводят индивидуально, с использованием источников УФ излучения. Стимулирующее действие УФ излучения направлено на синтез витамина Д, который участвует в процессах минерального обмена и окостенения.
- Водо- и теплолечение. При ПМД активно применяют местные ванны, хлоридно-натриевые, углекисло-сероводородные, радоновые ванны, а также грязелечение.
- Парафино-озокеритовые аппликации рекомендуются при контрактурах суставов и при нарушенных двигательных функциях. Теплолечение улучшает кровоснабжение мышц, повышает их эластичность. Температура озокерита 42 — 43 градуса, продолжительность 12 — 20 минут, до 10 процедур в день.
- Грязевые аппликации проводят на сегментарно-рефлекторную зону при температуре грязи 38 градусов. Под влиянием грязевых аппликаций улучшается кровоснабжение конечностей, повышается температура кожи на кистях рук и стопах.
Аппаратная физиотерапия
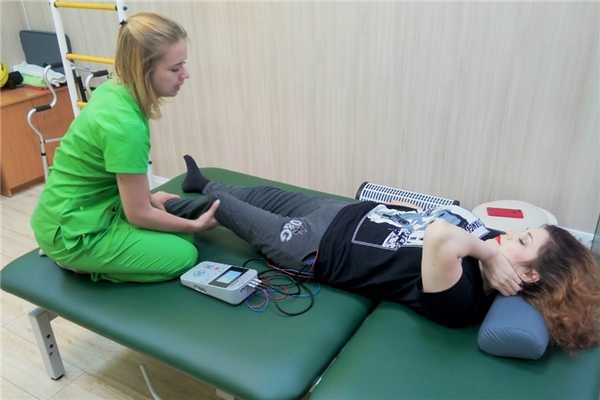
Среди аппаратных методов лечения приоритет отдают методам электролечения. Причина понятна, потому что основная потребность при ПМД — стимуляция мышечной активности. Но при этом, при назначении методик электролечения необходимо быть предельно осторожным, и внимательно следить за состоянием мышц и их ответом на электростимул.
Электростимуляция преимущественно проводится методикой СМТ. Воздействию подвергаются наиболее важные для статики мышцы. Силу тока подбирают индивидуально, с учётом указанного выше фактора настороженности. Сила тока выбирается минимальная, достаточная для того, чтобы вызвать ответное мышечное сокращение. Процедура продолжается 1 — 2 минуты, потом чередуется с 2 — 3 минутами отдыха.
Для введения лекарственных препаратов активно назначают процедуру лекарственного электрофореза. Наиболее часто применяют лекарственный электрофорез по методике ионного рефлекса, эндоназальный электрофорез витамина В, электрофорез прозерина.
- Лекарственный электрофорез по методу ионного рефлекса улучшает вегетативную регуляцию.
- Эндоназальный электрофорез витамина В применяется с 3 — 5 лет и способствует ускоренному всасыванию витамина В через слизистые оболочки носа.
- Электрофорез прозерина проводят по методике общего воздействия, можно сочетать с курсом диадинамотерапии.
Противопоказано применение электрофореза на область поражённых мышц, при наличии контрактур, при тяжёлой степени двигательных нарушений, а также при непереносимости назначенных препаратов.
Методы физической терапии применяют в самые начальные периоды болезни, как только установлен диагноз. Эффективность её больше при лёгкой и средней тяжести. Физиотерапия назначается в комплексе с лечебной физкультурой, массажем, лечебными препаратами, ортопедическими мероприятиями.
Ортопедические мероприятия при ПМД
Ортопедические мероприятия направлены на предупреждение и исправление деформаций, сохранение двигательной активности и подвижности конечностей. Предупреждение и консервативное исправление контрактур производят с помощью мышечных повязок и укладок шин. На разных стадиях болезни ортопедические укладки, посадки больного с фиксирующими повязками позволяют предупредить развитие деформаций. В процессе реабилитации больных ПМД используют специальные протезно-ортопедические приспособления, фиксирующие мышцы конечностей, что помогает сохранить опороспособность.
При невральной амиотрофии больному ребёнку изготавливают специальную обувь, фиксирующую стопу. Сохранение опороспособности конечностей имеет важное значение для физического и психического развития.
Помимо указанных выше методик медицинской направленности, детям также активно проводят психологическую реабилитацию. Это и трудотерапия, которая в детском возрасте проводится в форме игры. В процессе вовлечения ребёнка обучают базовым навыкам самообслуживания. Музыкотерапия также проводится в различных формах и по мере возможности, потому что не всегда есть возможность проводить занятия подобного рода в стационарах. Занятия музыкой расширяют возможности художественного и культурного развития, общения со сверстниками, настраивают на позитивный лад. Также под музыку хорошо проходят двигательные упражнения, что позволяет сочетать занятия с описанной выше игровой терапией.
Заключение
Исследования показали, что эффективность восстановительного лечения на всех этапах выше, если оно начато раньше. К сожалению, так получается далеко не всегда. А восстановление и поддержание двигательных функций получается гораздо лучше в более раннем возрасте и при менее запущенном процессе.
Об улучшении состояния больного свидетельствует:
- улучшение двигательной активности;
- повышение выносливости мышц;
- усиление сухожильных рефлексов;
- положительные сдвиги данных томографии, миографии, реовазографии, функциональных показателей.
Социально-трудовая реабилитация детей, не имеющих трудового опыта, заключается в профессиональном обучении. При выборе профессии учитывают медицинские, психологические и социально-профессиональные факторы.
Прогноз жизни определяется быстротой прогрессирования дистрофических изменений в скелетной мускулатуре, а также возрастом, при котором началась болезнь. Родителям, имеющим ребёнка с ПМД, нужно тактично рассказать о течении болезни, возможных последствиях. Но в то же время ни в коем случае нельзя отнимать надежду на определённый успех, возможность стабилизации, длительной ремиссии и создать более оптимистичную обстановку.
ЗАПИСАТЬСЯ НА КОНСУЛЬТАЦИЮ В НОВОСИБИРСКЕ:

Врач физиотерапевт, остеопат, реабилитолог Карасенко В.П. Городская Клиническая больница №25 города Новосибирска.
Прогрессирующая мышечная дистрофия Эрба-Рота
Прогрессирующая мышечная дистрофия Эрба-Рота — аутосомно-рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, т. е. начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений. Диагностика осуществляется методами неврологического осмотра, электрофизиологического тестирования, генеалогического исследования и генетического анализа, по показаниям проводится гистологический анализ биоптата мышечной ткани. Лечение только симптоматическое, позволяющее лишь продлить двигательную активность пациентов. Итог заболевания — полная обездвиженность.
Общие сведения
Прогрессирующая мышечная дистрофия Эрба-Рота - самый полиморфный вариант наследственной миодистрофии. От других видов прогрессирующей мышечной дистрофии вариант Эрба-Рота отличается вариабельностью как времени дебюта заболевания, так его клинической картины и течения. Заболевание описано в 1882 г. немецким неврологом Вильгельмом Эрбом. Одновременно в России изучением этой патологии занимался В.К. Рот, для обозначения миодистрофии он ввел термин «мышечная сухотка». В современной мировой и отечественной неврологии в честь этих исследователей употребляется название «прогрессирующая мышечная дистрофия Эрба-Рота». Частота встречаемости заболевания находится в пределах от 1,5 до 2,5 случаев на 100 тыс. населения.
Причины миодистрофии Эрба-Рота
Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов. На сегодняшний день генетике известно не менее 9 хромосомных локусов, аберрации в которых приводят к развитию миодистрофии Эрба-Рота. Чаще всего наблюдаются мутации в локусах 15q15-q21.1, 13q.
Около 30% генных аномалий возникают de novo, остальные носят семейный характер. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно. Болеют как мальчики, так и девочки. Патология проявляется, если ребенок получает аномальный ген от каждого из родителей. В случае, когда оба родителя выступают носителями аберрантного гена, но сами не болеют, вероятность развития миодистрофии у ребенка составляет 25%.
Симптомы миодистрофии Эрба-Рота
Прогрессирующая мышечная дистрофия Эрба-Рота манифестирует в среднем в возрасте 13-16 лет. Однако известны отдельные случаи дебюта болезни в раннем детском возрасте и в возрасте после 20 лет. Мышечная слабость и атрофии возникают в первую очередь в мускулатуре тазового пояса и проксимальных отделов ног. Отмечаются затруднения ходьбы по лестнице, подъема из положения присев на корточки. Типичен симптом Говерса — если пациент сидел на полу, то для того, чтобы подняться, он использует собственное тело как опору.
Со временем дистрофические изменения распространяются на мышечные группы туловища и рук. Такой тип распространения миодистрофии носит название восходящий. Он наиболее типичен для большинства наследственных мышечных дистрофий. Однако в некоторых случаях дистрофии Эрба-Рота наблюдается нисходящий тип распространения патологического процесса, когда мышечная слабость возникает вначале в руках, затем в тазовом поясе, а через несколько лет в мышцах ног.
Тотальная гипотрофия мышц туловища приводит к тому, что у пациентов начинают выступать лопатки (т. н. «крыловидные лопатки»), талия становиться очень тонкой (т. н. «осиная талия»), усиливается поясничный лордоз, живот выпячивается вперед. Характерен симптом свободных надплечий — при попытке приподнять больного вверх, удерживая его за подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними. Поражение лицевых мышц влечет за собой гипомимию (т. н. «лицо сфинкса»), неполное закрытие век, выворачивание и утолщение губ.
Миодистрофия Эрба-Рота сопровождается ранним угасанием коленных рефлексов, а также сухожильных рефлексов с плечевого бицепса и трицепса. Чувствительная сфера не нарушена. Псевдогипертрофии не столь характерны, как при мышечной дистрофии Беккера. Могут наблюдаться ретракции сухожилий и мышечные контрактуры, но они выражены в меньшей степени, чем аналогичные проявления мышечной дистрофии Дрейфуса. Со временем атрофия и слабость дыхательных мышц приводит к появлению прогрессирующей дыхательной недостаточности, возникает опасность развития застойной пневмонии. Миодистрофический процесс в гладкой мускулатуре обуславливает снижение кишечной перистальтики с тенденцией к запорам. Поражение сердечной мышцы влечет за собой возникновение кардиомиопатии, нарушений ритма, сердечной недостаточности.
Диагностика миодистрофии Эрба-Рота
Миодистрофия Эрба-Рота диагностируется неврологом и генетиком при сопоставлении данных анамнеза (возраст дебюта заболевания, последовательность развития симптомов), неврологического статуса пациента, ЭФИ нервно-мышечной системы, генеалогических данных, результатов анализа ДНК и микроскопического исследования мышечной ткани. Дифференцировать миодистрофию Эрба-Рота приходится от других форм этого заболевания (прогрессирующей дистрофии Дюшенна, миодистрофии Дрейфуса и Беккера), дерматомиозита, полимиозита, бокового амиотрофического склероза, токсической миопатии и др.
При неврологическом осмотре обращает на себя внимание снижение мышечной силы в мускулатуре проксимальных отделов ног и рук, гипотония и гипотрофия указанных мышц, гипорефлексия или полное выпадение локтевых и коленных рефлексов, сохранность всех видов чувствительности. ЭМГ и ЭНГ свидетельствуют о первичном поражении мышечной ткани при сохранности проведения импульсов по нервным стволам.
Генеалогическое исследование подтверждает аутосомно-рецессивный характер наследования. Исследование ДНК может выявить наличие генных мутаций. Однако отрицательный результат исследования не опровергает диагноз, поскольку не всякая мутация может быть обнаружена. Отрицательный анализ ДНК является показанием к биопсии мышц. В биоптате обнаруживаются различные по толщине мышечные волокна, уменьшенное число мышечных ядер, некротические и склеротические изменения.
Резкое повышение уровня креатинфосфокиназы характерно для начального периода дистрофии Эрба-Рота, затем происходит постепенное понижение этого показателя вплоть до нормальных цифр. Обзорная рентгенография грудной клетки позволяет выявить расширение границ сердца, наличие воспалительных изменений легочной ткани. ЭКГ зачастую определяет аритмию и нарушения проводимости. При помощи УЗИ сердца можно диагностировать кардиомиопатию. Для оценки степени сердечных нарушений требуется консультация кардиолога, при подозрении на пневмонию — консультация пульмонолога.
Лечение миодистрофии Эрба-Рота
Этиопатогенетическая терапия пока не разработана. Симптоматическое лечение направлено на как можно более длительное сохранение двигательной способности пациента. С этой целью применяют медикаментозные курсы, включающие АТФ, витамины Е и группы В, тиоктовую кислоту и др. Занятия лечебной физкультурой должны проводиться ежедневно и включать упражнения на все группы мышц. Регулярно назначаются курсы массажа и физиопроцедуры.
При поражении сердечной мышцы рекомендован инозин, сердечные гликозиды, антиаритмики. При развитии контрактур может потребоваться ортопедическое лечение. Выраженное снижение жизненной емкости легких из-за атрофии дыхательных мышц служит показанием к ИВЛ.
Прогноз и профилактика
Мышечная дистрофия Эрба-Рота может иметь различную тяжесть и скорость прогрессирования, что бывает выражено даже в пределах одной семьи. Описаны тяжелые дюшенноподобные варианты заболевания с ранним летальным исходом от дыхательной недостаточности, инфекционных поражений легких или сердечной недостаточности. В относительно легких вариантах миодистрофия может протекать без поражения сердечной мышцы, обездвиженность больных наступает лишь к 50-летнему возрасту. Профилактикой является своевременное генетическое консультирование семейных пар, планирующих зачатие ребенка; исключение близкородственных браков, в которых оба супруга могут стать носителями патологического гена.
Прогрессирующая мышечная дистрофия Беккера
Прогрессирующая мышечная дистрофия Беккера — вариант наследственной сцепленной с Х-хромосомой миодистрофии, отличающейся более замедленным и доброкачественным течением. Заболевание характеризуется постепенно усугубляющейся и распространяющейся мышечной слабостью, гипотонией и атрофией, первоначально возникающей в мышцах бедер и тазового пояса. Диагностический поиск включает неврологическое обследование, консультацию генетика и кардиолога, нейрофизиологическое тестирование нервно-мышечного аппарата, ДНК диагностику, биопсию мышц с морфологическим, иммунологическим и гистохимическим изучением полученных образцов. Лечение симптоматическое и, к сожалению, малоэффективное. Прогрессирование болезни приводит к потери больными способности самостоятельно передвигаться к возрасту 40 лет.
Прогрессирующая мышечная дистрофия Беккера впервые была описана в 1955 г. как доброкачественный вариант течения мышечной дистрофии Дюшенна. В последующем многочисленные исследования в области клинической неврологии, генетики и биохимии обнаружили существенные отличия в характере течения, биохимической и морфологической основе этих заболеваний. В результате клиническая форма Беккера была выделена как самостоятельная нозология.
Мышечная дистрофия Беккера входит в группу миопатий (миодистрофий) — заболеваний, возникающих вследствие нарушений строения и метаболизма мышечной ткани и проявляющихся мышечной слабостью. Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому болеют только лица мужского пола. Частота встречаемости составляет 1 новорожденный на 20 тыс. детей.
Причины
В основе заболевания лежит мутация в гене, ответственном за кодирование белка дистрофина. Примерно 30% от общего числа случаев мышечной дистрофии Беккера приходится на т. н. «свежие» мутации. Ген располагается в 21 локусе (в регионе Хр21.2-р21.1) короткого плеча Х-хромосомы. Примерно у 65-70% больных обнаруживаются крупные делеции указанного участка, у 5% - дупликации, у остальных — точковые мутации. Указанные структурные перестройки гена не влекут за собой полного прекращения синтеза дистрофина, как при дистрофии Дюшенна, а потенцируют синтез аномального усеченного белка, в некоторой степени способного выполнять свои функции. Это и обуславливает более доброкачественный характер дистрофии Беккера в сравнении с вариантом Дюшенна.
Патогенез
В норме белок дистрофин поддерживает целостность сарколеммы - мембраны миоцитов (мышечных волокон), обеспечивает эластичность и устойчивость миофибрилл при мышечном сокращении. Неспособность аномального дистрофина адекватно выполнять эти функции приводит к нарушению целостности мембран мышечных волокон. В следствие этого происходят дегенеративные изменения цитоплазматических компонентов последних и повышенная транспортировка ионов калия внутрь миоцитов. Результатом таких биохимических и морфологических сдвигов является гибель миофибрилл и разрушение мышечных волокон. На месте погибших миоцитов происходит образование соединительной ткани, что обуславливает феномен псевдогипертрофии — увеличение объема и плотности мышцы при резком снижении ее сократительной способности.
Симптомы
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10 до 15 лет, в некоторых случаях раньше. Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость в тазовом поясе и нижних конечностях. У ряда пациентов первыми проявлениями являются периодические болезненные мышечные судороги (крампи), локализующиеся в ногах. Мышечная слабость обуславливает затруднение при подъеме по лестнице, при необходимости встать из положения сидя. Со временем формируется переваливающаяся «утиная походка». Для того, чтобы встать, пациент вынужден использовать вспомогательные миопатические приемы — опираться руками о расположенные рядом предметы мебели или, при отсутствии таковых, использовать в качестве опоры собственное тело (симптом Говерса).
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично развивающимися атрофиями мышц. В первую очередь поражаются мышцы бедра и тазового пояса, затем процесс распространяется на мускулатуру плечевого пояса и проксимальных мышц рук. В начале болезни формируются псевдогипертрофии, наиболее выраженные в икроножных, дельтовидных, трех- и четырехглавых мышцах. По мере прогрессирования миодистрофии они трансформируются в мышечные гипотрофии.
Клиническая картина мышечной дистрофии Беккера во многом сходна с миодистрофией Дюшенна. Усугубление мышечной слабости с течением времени приводит к обездвиженности пациента и формированию контрактур суставов. Однако развитие дистрофического процесса в мышечной ткани при дистрофии Беккера идет гораздо медленнее, что обуславливает длительную двигательную активность больных. В среднем пациенты сохраняют способность самостоятельно передвигаться до 35-40-летнего возраста. Кроме того, дистрофия Беккера не сопровождается олигофренией, выраженным искривлением позвоночника и другими скелетными деформациями. Возможна кардиомиопатия дилятационного или гипертрофического типа, блокада ножек пучка Гисса, но сердечно-сосудистые расстройства выражены умеренно. Может наблюдаться снижение либидо, гинекомастия, атрофия яичек, импотенция.
Диагностика
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных, дополнительных обследований и генетического тестирования. В неврологическом статусе наблюдается снижение мышечной силы и умеренное снижение мышечного тонуса в проксимальных отделах конечностей, выпадение коленных рефлексов при симметричном снижении сухожильных рефлексов дистальных отделов ног и верхних конечностей, полная сохранность чувствительности.
Среди клинических анализов наибольшее значение имеет биохимический анализ крови, который выявляет многократное повышение уровня КФК. Данные электронейрографии позволяют исключить поражение нервных волокон, электромиография свидетельствует о первично-мышечном типе поражения. Биопсия мышц проводится только после отрицательных результатов генетического анализа. Морфологическое исследование полученного материала определяет диффузную разнокалиберность, дистрофические и некротические изменения мышечных волокон, разрастание соединительной ткани. Проводится специальное иммунное окрашивание образцов с последующим определением наличия в них дистрофина.
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа ДНК. Выявление дупликаций или делеций в гене Хр21 дает возможность установить точный диагноз. Отрицательный результат анализа ДНК не говорит об отсутствии патологии, поскольку могут иметь место точковые мутации, поиск которых представляет собой сложную и более дорогостоящую процедуру.
С целью выявления сердечной патологии назначается электрокардиография, Эхо-КГ, консультация кардиолога. Кардиологическое обследование может обнаружить нарушение внутрижелудочковой проводимости, АВ-блокаду, дилатацию желудочков, гипертрофические изменения миокарда, кардиомиопатию, сердечную недостаточность.
Пренатальная диагностика рекомендована, когда мать является носителем патогенного гена. Если ребенок мужского пола, то вероятность развития заболевания у него составляет 50%. Биопсия хориона может проводиться в сроке 11-14 нед. беременности, амниоцентез — после 15-й недели, забор пуповинной крови (кордоцентез) — на сроке больше 18 нед.
Дифференциальная диагностика
Дифференциальная диагностика проводится с прогрессирующей мышечной дистрофией Дрейфуса, миодистрофией Дюшена, мышечной дистрофией Эрба-Рота, метаболической миопатией, полимиозитом и дерматомиозитом, воспалительной миопатией, спинальной амиотрофией, наследственной полиневропатией.
Лечение миодистрофии Беккера
На современном этапе несколькими группами ученых ведутся настойчивые исследования в области поиска эффективных методов лечения прогрессирующих миодистрофий. В настоящее время пациенты получают в основном метаболическую и симптоматическую терапию. Разработаны различные схемы лечения, позволяющие улучшить двигательные возможности больного и несколько замедлить прогрессирование болезни.
Терапия глюкокортикоидами используется для снижения скорости прогрессирование атрофии мышечной ткани. Пациентам назначают метаболические средства (витамины группы В, левокарнитин), витамин D и препараты кальция для профилактики остеопороза, β-адреноблокаторы и ингибиторы АПФ для предупреждения кардиомиопатии, диуретики при сердечной недостаточности.
Наблюдения показали, что постельный режим усугубляет мышечную слабость. Поэтому пациентам рекомендуется умеренная физическая активность, занятия плаванием. Поддержание мышечной эластичности и силы, а также профилактика контрактур проводится средствами массажа, физиотерапии и лечебной гимнастики. Применение различных ортопедических средств (ходунков, инвалидных колясок, фиксаторов для ног, экзоскелетов) позволяет расширить двигательные возможности пациентов и их способность к самообслуживанию. По показаниям проводится хирургическое лечение контрактур.
Прогноз и профилактика
Прогрессирующая мышечная дистрофия Беккера имеет неблагоприятный прогноз. Хотя обездвиженность у пациентов наступает гораздо позже, чем при дистрофии Дюшенна, в конечном итоге поражение сердечной мышцы и дыхательной мускулатуры приводят к гибели пациентов от сердечной или дыхательной недостаточности. Продуманный уход, адекватная терапия, вентиляционная поддержка дыхания, применение ортопедических средств могут лишь увеличить продолжительность и улучшить качество жизни пациента. Профилактика заключается в предупреждении рождения ребенка с патологией путем генетического консультирования будущих родителей и проведение пренатальной диагностики.
Прогрессирующая мышечная дистрофия Дюшенна
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Миодистрофия Дюшенна и пути реабилитации

Татьяна Андреевна Гремякова, президент благотворительного фонда «Гордей» составила пошаговую инструкцию для родителей мальчиков с миодистрофией Дюшенна
Развитие орфанного генетического заболевания миодистрофии Дюшенна проходит четыре стадии. В зависимости от них врачи назначают пациентам различные виды реабилитации. На что обратить внимание на разных стадиях течения болезни и как помочь пациенту читайте в руководстве, подготовленном по итогам первой всероссийской онлайн-конференции по миодистрофии Дюшенна. Это мероприятие организовал Благотворительный фонд «Гордей».
Важно провести тестирование ребенка по шкалам Бейли. Это поможет определить задержки в различных областях развития: физической, когнитивной, коммуникативной. Определив проблему, можно начать ранние корректирующие вмешательства. К ним относится не только физическая терапия, но и помощь логопеда или психолога.
В своем выступлении профессор и реабилитолог Имельда Де Грут (Нидерланды) отметила, что важно проверять на МДД и детей с задержками развития. Заболевание связано не только с мышцами, но и с мозгом. Связано это с определенной изоформой гена дистрофина в мозге. Если есть ее дефицит, то возможны интеллектуальные и когнитивные проблемы.
У мальчиков с Дюшенном уровень IQ в среднем на одно стандартное отклонение ниже нормы, 27% пациентов имеют сниженный IQ. У большей части мальчиков с Дюшенном — нормальный и высокий уровни IQ. Некоторые мальчики успешно заканчивают университеты и получают ученые степени. Поэтому нельзя по умолчанию воспринимать пациента с Дюшенном как человека с задержкой когнитивного развития.
Миодистрофия Дюшенна: что это такое? Как часто и почему возникает это заболевание, почему педиатры нередко путают его с гепатитом, что делать, если вашему ребенку поставлен такой диагноз
В медицинском аспекте нужно обратить внимание:
- На растяжки для профилактики контрактур голеностопного сустава (желательно, чтобы родители ежедневно делали ребенку растяжки в форме игры, а не «лечения»). В этот период ребенку также прописывают кортикостероиды, в ночное время начинают использовать туторы.
- На нарушения, не имеющие отношения к мышцам (речевая терапия, помощь логопеда, когнитивное обучение, помощь поведенческого терапевта).
- На слабость мочевого пузыря — 83% пациентов сталкиваются с проблемами (капельное подтекание мочи, натуживание, неполное опорожнение). Из-за этого, став старше, дети стараются меньше пить, так как им не хочется часто ходить в туалет. При этом, сев в инвалидное кресло, ребенок начинает принимать диуретики. В этой связи могут возникать проблемы с почками.
- На физическую активность. Ребенку нужно дать почувствовать, что такое прыгать и карабкаться по лестнице, что такое «высоко-низко» и «быстро-медленно», это поможет ему научиться считать.
В социальном аспекте на ранней амбулаторной фазе важно:
- Чтобы ребенок играл со сверстниками, заводил друзей. Обычно, друзья, появившиеся на этом этапе, остаются с ним и далее.
- Выбрать школу. Хотелось бы, чтобы дети ходили в обычную школу, это важно для их интеграции в обществе. Но если у ребенка целый букет проблем, в том числе и когнитивных, то выбор школы - более трудная задача.
- Не забывать о дислексии — проверить ребёнка до начала обучения чтению и, в случае выявления, обсудить это с учителем и соответствующим образом адаптировать программу обучения.
- Обсудить с учителем особые нужды ребенка. Если ребенок в классе говорит, что хочет пойти в туалет, то ему нужно дать эту возможность. Чтобы не возникало ситуаций, когда учитель говорит: «Нет, подождешь до конца урока». Ведь мальчик с Дюшенном не может ждать.
- Выбрать спорт, которым можно заниматься долго. Например, плавание. В воде не чувствуется веса и ребенок может двигаться, как ему хочется. При этом нет риска падения или избыточной нагрузки на мышцы. В Голландии используют боевые искусства. Они учат правильно падать и продолжают заниматься даже в инвалидном кресле. Например, здоровый ребенок может играть на мягких мечах с ребенком с Дюшенном. Можно присмотреться к видам спорта, в которые играют на инвалидной коляске: футбол, хоккей. Даже если пока ребенок ходит самостоятельно. Главное, чтобы у мальчика возникла ассоциация, что спорт - это весело.
- На этом этапе важно поддерживать здоровье костей: принимать витамин D и кальций.
- Увеличивается опасность падений и переломов (особенно длинных трубчатых костей), при падении назад (на копчик, на спину) могут возникать переломы и трещины в позвонках. Поэтому если ребенок жалуется на боль в спине, нужно обязательно провести рентгенологическое исследование для исключения возможности перелома.
- Снижается физическая активность, выносливость. Некоторые ребята ощущают, что слабеют руки: им тяжело дотягиваться до предметов или проводить действия, требующие выносливость мышц рук. Они меньше ходят, меньше двигаются, при падении им тяжело подняться. Дети начинают избегать хождения. В результате возникают проблемы с кишечником (запоры), поскольку всё больше замедляется его перистальтика.
- На этой фазе мышечная масса, сила и выносливость снижаются. Появляется проблема дисфункциональной атрофии. Тут нужны тренировки рук и ног. В Голландии используют специальные электротренажеры и подставки под руки во время еды и пребывания за компьютером. Конечно, они не изменят течение заболевания, но с их помощью можно избавиться от проблемы дисфункциональной атрофии.
- Пора начать использовать шины/туторы/ортезы для ног в ночное время. Они бывают статические (четко держат стопу в определенном положении) и динамические (при помощи специального приспособления на шине можно растягивать ахиллово сухожилие). В дальнейшем потребуются и шины для рук. У всех Дюшеннов способность к супинации снижается и происходит локтевое отведение кисти, в результате чего пальцы все более изгибаются (скрючиваются). Поэтому одну ночь шину надевают на правую ручку, на следующую ночь — на левую, чтобы у ребенка была возможность подать сигнал тревоги или самостоятельно что-то сделать свободной рукой.
К каким изменениям нужно подготовиться в социальном аспекте:
- Нужно начать готовить ребенка к тому, что в дальнейшем он будет передвигаться в кресле.
- Родителям нужно задуматься об изменении дома. Например, приступочки на ступенях лестницы или стул в душе, помогут ребенку преодолеть боязнь падения. На улице можно использовать беговелы, чтобы мальчик не переносил вес всего тела, но при этом тренировал ноги.
На что важно обратить внимание в медицинском аспекте:
- При адаптивных тренировках полезна гидротерапия. Она тренирует оставшиеся функционирующие мышцы. Важно, чтобы в течение всего дня ребенок оставался активным. Подойдут специальные тренажеры для домашнего использования. Например, электрические колеса у кресла. Для передвижения их нужно крутить руками, что сохраняет постоянную активность. Можно использовать опоры для поддержки рук. Но у них высокий кронштейн и при размещении на коляске могут возникнуть проблемы. Есть электроприводные и механические вспомогательные устройства поддержки рук. Но эксперты считают, что будущее за экзоскелетом. В ближайшее время портативные экзоскелеты пройдут тестирование на длительное ношение в домашних условиях.
- В кресле-коляске мальчику должно быть комфортно и безопасно. Используйте вспомогательные крепления, если есть риск падения из кресла.
- Обязательно нужно работать над предотвращением развития эквиноварусной деформации стопы.
- Ассиметричная контрактура вокруг малого таза может приводить к явлению «перекошенного таза». Позвоночник все более искривляется, и в результате развивается сколиоз. Если вы видите, что ребенок сидит ассиметрично в коляске, подумайте, возможно ли с этим что-то сделать.
- Подростки и молодые взрослые с Дюшенном сталкиваются с проблемой боли в спине, бедрах и частой потребностью менять положение. Хороший вариант — коляска с возможностью отклониться назад-вперед. Также они жалуются на усталость при сидении и на давление на кожу. Поэтому им необходимо находить оптимальное положение.
- Если ребенок оказывается в коляске, важно использовать правильную обувь. Она должна быть достаточно высокой и устойчивой на подставке для ног. Кроме того, важно наличие у коляски боковых поддерживающих подставок, чтобы ноги не «разваливались» в позу «лягушки».
- Голова и спина при нахождении в коляске должны быть правильно расположены. Этому помогут специальные устройства для индивидуального позиционирования. Если есть сколиоз, важно поддерживать прямой позвоночник. Если таз перекошен — важно сделать ассиметричной подушку, на которой сидит пациент, чтобы у него была возможность держать спину прямой. Нужно понимать, что кресло подбирается индивидуально и должно соответствовать анатомическим особенностям конкретного человека.
- После 14 лет важно исследовать ребенка по шкале оценки стадии полового созревания Таннера , чтобы определить необходимость приёма тестостерона или снижения дозировки кортикостероидов (при необходимости).
На что надо обратить внимание в медицинском аспекте:
- Когда здоровый человек ест, то сначала разжевывает пищу, затем язык подталкивает её к гортани, а потом пища проталкивается в пищевод. В этот момент дыхательные пути закрываются своеобразной «крышечкой». Если подъем этой «крышечки» невозможен, то дыхательные пути закрываются не оптимально, при ослабленных мышцах гортани еда может застрять и не дойти до пищевода.
- На поздней неамбулаторной стадии возникают проблемы с глотанием (особенно твердой пищи), может затрудняться процесс жевания. Поэтому врачи рекомендуют более жидкое питание. Нужно пить воду после еды, чтобы протолкнуть еду и предотвратить попадание пищи в легкие. При необходимости назначается питание через гастростому.
Социальный аспект поздней неамбулаторной фазы:
Согласно опросу 213 пациентов до 35 лет, в поздней неамбулаторной стадии никто из них не работает, не занимается спортом, у них нет хобби. Только у нескольких опрошенных были романтические отношения. В Нидерландах есть несколько мужчин, которые женились и завели детей, но их процент среди всех пациентов — пока невелик.
При постановке диагноза ребенку у родителей рушится мир. Поэтому врачу важно подробно ещё раз всё разъяснить, ответить на все вопросы. Нужно акцентировать внимание родителей на том, как они смогут улучшить качество жизни ребенка, несмотря на то, что в будущем ему придется пользоваться инвалидным креслом.
Я верю, что скоро мальчики с Дюшенном будут жить не так, как сейчас Личная история семьи Татьяны Андреевны Гремяковой, президента благотворительного фонда «Гордей»
- Заботьтесь о себе. Родителям пациентов с Дюшенном непрерывно приходится решать вопросы, о которых другие даже не задумываются: дополнительная финансовая нагрузка, будущее ребенка и удовлетворение его потребностей. А ведь нужно заботиться не только о ребенке с Дюшенном, но и об остальных членах семьи. Так возникает ощущение, что нет выхода. Поэтому родителям важно заботиться и о себе. Ведь если подорвано физическое или эмоциональное здоровье взрослых, то тяжело обеспечить надлежащий уход и отвечать потребностям ребенка.
- Совет папам детей с Дюшенном. Вовлекайтесь в уход за ребенком, ходите с ним к врачу. Не используйте работу как средство избегания семьи , ухода от реальности. Да, для обеспечения потребностей сына важна финансовая стабильность, чтобы обеспечить ему уход и медицинскую помощь. Но не используйте это в качестве предлога, чтобы, уйдя на работу, оставить стресс дома. Не делайте этого. Иначе мама выгорит.
- Вкладывайте силы и время в отношения со своим партнёром . Отчуждение развивается очень быстро. Отношения — это работа. Нужно проводить время с супругом /супругой. Беречь романтические отношения.
- В дружеские отношения тоже нужно инвестировать время и хотеть, чтобы они развивались. Мы с женой, когда узнали диагноз сына, потеряли часть знакомых. Но были и те, кто остался рядом и всегда нас поддерживал. Появились новые друзья из числа семей пациентов с Дюшенном.
- Когда вам нужна помощь родных или друзей просите о ней прямо, с четко сформулированными потребностями. Люди не умеют читать мысли. Они не отстраняются и хотят помогать, только об этом нужно говорить, даже если вы чувствуете себя неуютно.
- Важная вещь для баланса в жизни — хобби. С его помощью вы поймете, что не всё в жизни связано с работой или с Дюшенном. Родитель мальчика с МДД — это не весь вы. Есть другие части жизни. Пусть в вашей жизни не всё определяет заболевание ребенка.
- Старайтесь видеть хорошее даже в сложных ситуациях, всюду ищите повод для улыбки. Понятно, что тяжело радоваться, если у вас ребенок с Дюшенном. Но нужно искать положительные моменты, чтобы отрицательные стороны заболевания не поглотили вас целиком.
- Будьте осторожны с соцсетями . Они, конечно, могут быть важным источником поддержки, рекомендаций и обмена информацией. Но могут оказывать и отрицательное воздействие. Люди бывают грубы, иногда ведут себя неадекватно, бывают критичны. Важно контролировать, чтобы соцсети вас не поглотили.
- Старайтесь уделять внимание другим детям в семье. Вовлекайтесь в их жизнь, чтобы они себя чувствовали хорошо. Наша 13-летняя дочь часто бывала с нами на приемах у докторов с братом. В какой-то момент она сказала: «Вот бы у меня тоже была миодистрофия Дюшенна!». Нас это шокировало и надо было воспринять этот тревожный звоночек, чтобы такого больше не происходило.
- Иногда бабушки и дедушки могут вас не понимать. При появлении поведенческих проблем у внука с МДД, старшие винят родителей, что они балуют ребенка, неправильно его воспитывают. Это, конечно, вызывает чувство раздражения и досады. Поэтому нужно пояснить им неврологическую основу поведения вашего ребенка. Тогда вас поймут.
Читайте также: