Синдром Адамса-Кершнера (Adams-Kershner) - синонимы, авторы, клиника
Добавил пользователь Cypher Обновлено: 21.01.2026
ГБОУ ВПО «Дагестанская государственная медицинская академия» Минздрава РФ, Махачкала
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2015;115(9): 49‑53
Синдром Хакима—Адамса — малоизвестное, хотя и достаточно часто встречающееся состояние. Этиология его не полностью ясна. Патогенетические механизмы включают блокаду оттока цереброспинальной жидкости (ЦСЖ) по верхнелатеральной поверхности мозга, развитие гидроцефалии с расширением желудочков мозга при сохранении нормотензии ЦСЖ. Основные клинические проявления: развитие атаксии, апраксии, явления деменции и нарушения мочеиспускания, вплоть до недержания мочи. Диагностика основывается на данных клинической симптоматики, люмбальной пункции с пробным извлечением жидкости и нейровизуализации. Основным способом лечения является наложение вентрикулоперитонеального шунта для отведения ЦСЖ. Своевременное проведение хирургического вмешательства дает хорошие результаты, улучшает качество жизни пациентов.
Синдром Хакима—Адамса (СХА), или нормотензивная гидроцефалия, описан в 1964—1965 гг. [1]. Он включает в себя классическую триаду симптомов: нарушение походки в результате атаксии или апраксии, или лобной дисбазии, деменцию и недержание мочи. Заболевание встречается редко, его часто принимают за ригидную форму болезни Паркинсона (БП), болезнь Альцгеймера (БА) и распознают с трудом [2].
За рубежом заболевание известно широко и оперативному лечению подвергнуты сотни тысяч больных. В России оно известно меньше [3].
Сведения о распространенности противоречивы. Наиболее часто СХА встречается после 60 лет. По данным D. Jaraj и соавт. [4], заболевание встречается в 0,2% случаев в возрасте 70—79 лет и в 5,9% — 80 лет и старше. Авторы полагают, что реальные показатели значительно выше. C. Iseki и соавт. [5] выявили, что в японской популяции заболеваемость лиц старше 70 лет равна 1,2 на 1000 человек в год. Различия между мужчинами и женщинами не отмечены.
СХА у взрослых может быть следствием субарахноидального и внутрижелудочкового кровоизлияния, черепно-мозговой травмы, воспалительного процесса, перинатального поражения головного мозга и мозговых оболочек, объемных интракраниальных образований (опухоли, аневризмы мозговых сосудов), аномалий развития мозга, перенесенных операций на головном мозге и других ситуаций, создающих механические препятствия нормальной циркуляции цереброспинальной жидкости (ЦСЖ) [3]. Однако в большинстве случаев болезнь развивается без видимых причин.
Представляют интерес данные о возможности участия генетических факторов в развитии болезни [6]. Ряд симптомов СХА встречался среди родственников с частотой 7,1%, тогда как в контрольной популяции — лишь у 0,7%.
Причина клинических проявлений СХА до конца неизвестна. Возможно, они обусловлены растяжением волокон лучистого венца головного мозга.
Вероятный патогенетический механизм, лежащий в основе заболевания, — дисбаланс секреции и резорбции ЦСЖ и нарушение ликвородинамики [7]. Основным местом резорбции ЦСЖ у человека являются конвекситальные субарахноидальные пространства в области верхнего сагиттального синуса. При СХА возникают блокада оттока ЦСЖ по верхнелатеральной поверхности мозга, а также затруднение ее всасывания в кровь. Происходит увеличение объема ликворного пространства (размеры желудочков) с соответственным уменьшением объема мозговой ткани [8]. Изменения в субарахноидальном пространстве предшествуют расширению желудочков [5]. Причиной таких явлений может быть окклюзия ликворных путей в пределах желудочков (окклюзионная форма) или нарушение резорбции ЦСЖ из субарахноидального пространства через арахноидальные ворсины в дуральные полости мозга, являющиеся основными путями оттока венозной крови из мозга (открытая форма). Как уже указывалось, окклюзия и нарушение резорбции жидкости могут вести к растяжению проводящих путей лучистого венца.
Для СХА характерно постепенное развитие симптоматики — нарушений походки (моторные изменения), признаков органического поражения мозга (деменция, потеря памяти, дезориентация) и тазовых расстройств (дизурия, недержание мочи) [3].
Моторные нарушения развиваются медленно, появляются затруднения при ходьбе, создается впечатление повышения тонуса мышц. Больные ходят медленно, мелкими шажками, походка становится шаркающей, семенящей, на широко расставленных ногах. Отмечаются плохой контроль равновесия, неустойчивость при поворотах. Теряется осанка, появляется сгорбленная поза.
Больные сообщают, что затруднения возникают из-за того, что появляется тяжесть в икроножных мышцах, ноги становятся «тяжелыми», больным трудно их поднимать. Вследствие этого ходьба ограничивается по длительности и дальности. Могут отмечаться постуральный тремор, проявления типа акинетико-ригидного синдрома (феномен «застывания»). Такие проявления сближают заболевание с ригидной формой БП (этот диагноз довольно часто выставляется первоначально). Однако при детальном обследовании ригидность мышц не выявляется. У ряда пациентов встречается псевдобульбарный синдром.
В большинстве случаев нарушения ходьбы являются первым симптомом, затем возникает деменция и позднее присоединяются тазовые расстройства.
Нарушения когнитивных функций в целом напоминают таковые при Б.А. Наиболее значительными могут быть выпадение как кратко-, так и долговременной памяти, дезориентированность во времени, реже они проявляются совместно. Пациенты с трудом излагают историю своей болезни [1]. Появляются проблемы в исполнительной функции: планировании, сосредоточении, абстрактном мышлении [9].
Возникают нарушения семантической памяти. Оскудевает эмоциональная сторона, появляются апатия, благодушие [10]. Возможны явления агнозии: нарушение различных видов восприятия (зрительное, слуховое, тактильное). Замедляется скорость психических процессов и психомоторных реакций. Эти явления считают характерными для дисфункции передних отделов головного мозга и подкорковой деменции [2, 11, 12]. Степень нарушений когнитивных функций различна. На ранних этапах изменения не столь выражены, однако при длительном течении болезни приближаются к таковым при БА.
Тазовые нарушения появляются в последнюю очередь. Однако при целенаправленном расспросе уже на ранних стадиях СХА удается выявить жалобы больных на учащенное мочеиспускание и никтурию. Постепенно к этим симптомам присоединяются императивные позывы к мочеиспусканию, а затем недержание мочи. У пациентов теряется запирательный рефлекс, и на фоне когнитивных нарушений они начинают индифферентно относиться к данному факту, что характерно для лобного типа тазовых расстройств.
Диагностика СХА является сложной задачей, поскольку он может имитировать многие другие неврологические заболевания. Так, начальные проявления болезни напоминают ригидную форму БП, а более поздние — Б.А. При детальном обследовании БП исключается более или менее легко [13]. Магнитно-резонансная (МРТ) и компьютерная томография (КТ) головного мозга дают возможность исключить Б.А. Дифференциальной диагностике помогает также то, что при СХА после шунтирования наступает регресс симптомов [14]. МРТ является методом выбора в диагностике гидроцефалии.
При МРТ головного мозга выявляют расширение III и боковых желудочков. Особенно значительно расширены III желудочек, височные и фронтальные рога боковых желудочков. Гидроцефалия при этом синдроме является сообщающейся, так как сильвиев водопровод проходим.
В диагностике используются также люмбальная пункция и исследование ЦСЖ. Давление ЦСЖ в спинномозговом канале бывает в пределах нормы (что отражает и название болезни — нормотензивная гидроцефалия). Начальное давление должно быть менее 180 мм вод. ст. Имеются также количественные методы оценки ЦСЖ [15], в том числе автоматизированные. Показано, что объем ЦСЖ при СХА ниже, чем при БА [16] и у здоровых.
Есть исследования, показавшие, что в ЦСЖ при этом синдроме снижается уровень простагландин D-синтазы, что может быть использовано в диагностике [17]. Иногда проводится цистернография с использованием радиоактивного 99-m технеция или другого изотопа. Она позволяет установить замедление всасывания ЦСЖ на верхнелатеральной поверхности мозга. Специфичность и чувствительность метода требуют уточнения, и результаты спорны, поэтому методика используется редко. Другие методы исследования (М-ЭХО-ЭГ и ЭЭГ) малоинформативны. Диагностическими критериями СХА считают увеличение переднерогового индекса (индекс Эванса) более 30% и расширение височных рогов боковых желудочков более 2 мм [3].
Одним из диагностических приемов является пробное извлечение 30—50 мл ЦСЖ. Пока окончательно не решено, когда и как оценивать результаты этого теста. В основном оценка производится через 1 сут. При СХА после такой процедуры походка и когнитивные функции временно улучшаются. Однако K. Kang и соавт. [18] описали пациента, у которого через 1 сут отклика не было, но улучшение наступило через 7 сут. Пациент был шунтирован с хорошим послеоперационным исходом.
Имеются единичные работы, посвященные поиску средств консервативного лечения болезни. Так, N. Alperin и соавт. [19] выявили, что малые дозы ацетазоламида уменьшали объем перивентрикулярной жидкости и у 5 из 8 пациентов привели к улучшению походки. Показано также, что психотропные препараты второго поколения оказывают положительное влияние на клинические проявления [20].
Основным способом лечения признается хирургический, заключающийся в выполнении ликворошунтирующих операций [21]. Рекомендуется проводить оперативное лечение сразу после установления диагноза [22].
Одним из наиболее часто применяемых методов является вентрикулоперитонеальное шунтирование. Проксимальный отдел катетера устанавливают в боковом желудочке мозга, а периферический — отводят в брюшную полость. Считается, что за 1 сут в брюшную полость может попасть около 300 мл ЦСЖ. Однако она не доставляет особых беспокойств и утилизируется путем всасывания брюшиной (или слизистой оболочкой).
Существует несколько вариантов шунтирующих систем. Имеется простой дренажный катетер. Более технологически совершенными являются специальные катетеры для шунтирования. Как правило, они снабжены клапанами, регулирующими отток в зависимости от количества жидкости в системе желудочков головного мозга и препятствующими обратному току жидкости из брюшной полости в головной мозг. Система для шунтирования включает также резервуар, позволяющий при необходимости забирать жидкость на исследование и вводить лекарственные вещества. Имеются исследования, продемонстрировавшие, что отдаленные результаты операции при применении разных шунтовых систем примерно одинаковы [23]. Ряд авторов [24, 25] находили преимущества у систем с гравитационным регулированием клапана шунта. Метаанализ работ по шунтированию показывает, что у адекватно диагностированных пациентов шунтирование приводит к длительным положительным результатам.
Исходы тем лучше, чем моложе пациент, чем меньше у него коморбидных заболеваний и нет поражения белого вещества [26]. Эффективность шунтирования выше при меньшей длительности заболевания. У больных с нарушениями походки улучшение наступает в 77% случаев. Однако проведенные контролируемые исследования методами актиграфии показали, что при этом активность пациентов значительно не возрастает [27]. M. Poca и соавт. [28] показали, что после шунтирования у 79,3% пациентов наступило улучшение походки, у 82,4% — тазовых нарушений и у 63,7% — уменьшение когнитивных расстройств.
Неврологические расстройства при СХА могут полностью или в значительной степени регрессировать после своевременно проведенной шунтирующей операции. По данным разных авторов [2], хирургическое лечение оказывается эффективным в 1/3 —¾ случаев. В работе, оценивавшей исходы через 1 год после шунтирования [29], было показано, что у 69% пациентов наступило значительное улучшение и клинических проявлений (способность жить самостоятельно), и рентгенологических данных. Стадия болезни и коморбидность не влияли на степень улучшения. Однако другие авторы [30] нашли, что бо́льшая коморбидность влияет на успех операции: при наличии до шести сопутствующих заболеваний шунтирование не приводит к улучшению.
Осложнения встречаются у 31—38% пациентов. Основными являются синдром ликворной гипотензии, проявляющийся головными болями при вставании, в редких случаях — субдуральная гематома вследствие быстрого уменьшения размера желудочков. По данным M. Poca и соавт. [28], смертность составляет 0,8%. Ранние послеоперационные осложнения были у 5,3% оперированных. Через 6 мес бессимптомные гигромы отмечены у 3,4%. В последующем у 3% выявлены субдуральные гематомы. Для профилактики осложнений рекомендуют индивидуальный подбор шунта.
Приводим краткие описания собственных наблюдений.
Пациент М., 74 года, работник умственного труда. Несколько лет назад появились затруднения при ходьбе, шаркающая походка. Нарушения прогрессировали, стало трудно ходить пешком на работу (примерно 1 км). В дальнейшем стали беспокоить частые позывы к мочеиспусканию. Недержания мочи не отмечает. Когнитивные нарушения не проявлялись, хотя стал отмечать некоторое снижение памяти, трудности сосредоточения при умственной работе.
Анализы: общие анализы крови, мочи в норме.
Глюкоза крови 5,99 ммоль/л, инсулин 2,37 мкед/мл, пролактин 285 мкг/л; Т3 свободный 12,6 нмоль/л; тиреотропный гормон 3,66 мкМЕ/мл; Nа + 148 ммоль/л; K + 4,8 ммоль/л; Ca ++ 1,16 ммоль/л, лактатдегидрогеназа 212 ЕД/л.
Консультация окулиста: глазное дно без застойных явлений. МРТ головного мозга: мозговые структуры не смещены, желудочки умеренно расширены. Боковые желудочки симметричны, несколько увеличены, увеличение симметричное. III желудочек 13 мм, боковые — 24—25 мм. В белом веществе очаги микроглиоза 3—6 мм, интенсивность сигналов повышена. Микроангиопатия. Визуализируется расширение периваскулярных пространств в области базальных вен. Субарахноидальное пространство умеренно расширено в конвекситальных областях лобно-теменных долей. Гипофиз 3—3,4 мм, не увеличен. Области интенсивных сигналов в его структуре не выявляются. Ножка гипофиза распределена срединно. Стволовые структуры без особенностей. Позвоночные артерии несколько асимметричны по калибру.
Заключение: единичные очаги микроглиоза в белом веществе головного мозга как проявления микроангиопатии. Умеренное расширение боковых и III желудочков, ликворных пространств в области сильвиевых щелей. Проявления атрофических изменений головного мозга.
Диагноз: СХА, ранняя стадия.
При люмбальной пункции повышения давления ЦСЖ не отмечено. Удалено 30 мл ЦСЖ. Анализ в норме. После пункции почувствовал облегчение при ходьбе.
Операция была проведена в НИИ нейрохирургии им. Н.Н. Бурденко: с правой стороны установлен шунт, проксимальный конец в IV желудочке, дистальный — в брюшной полости в подпеченочном пространстве. Степень устойчивости 90% по шкале Карновского. Операция прошла успешно, заживление ранок первичным натяжением. Самочувствие улучшилось, голова «прояснилась», ходить стало легче. Рекомендовано в течение 1 мес исключить физические нагрузки, работу, требующую наклона головы, туловища, затем постепенно расширять режим.
Состояние пациента значительно улучшилось. Переносимость физических нагрузок значительно увеличилась — может быть на ногах до 3 ч без признаков тяжести, усталости. Стал активнее физически и психически — улучшилась умственная деятельность, активно занимается научной деятельностью и преподаванием.
Пациент: А., 62 года. Болеет около 2 лет. Появились непонятная слабость, когнитивные нарушения — стал «заговариваться», снизилась память, возникли конфликты на работе, в связи с чем вынужден был уйти со службы. Постепенно возникли затруднения при ходьбе, появилась боль в икрах обеих голеней, походка стала семенящей, замедленной. Установлен диагноз паркинсонизма, ригидная форма. Проводилось лечение мадапаром, пронораном, ПК-мерц, но без эффекта. Отмечалось нарастание симптоматики.
При обследовании все анализы в норме.
МРТ: в обеих полушариях головного мозга определяются участки изменения интенсивности МР-сигнала, гиперинтенсивные на Т2 и FLAIR, гипоинтенсивные на Т1-взвешенных изображениях, без признаков объемного воздействия. Боковые желудочки мозга симметричные, передние рога умеренно расширены. III желудочек до 10 мм, IV — не изменен.
Субарахноидальные пространства и сильвиевы щели расширены. Хиазмально-селлярная область без особенностей. Гипофиз имеет однородную интенсивность сигнала. В придаточных пазухах носа определяется усиление сигнала. Миндалины мозжечка расположены на уровне линии Чемберлена. Зрительные бугры, зрительные и слуховые нервы не изменены.
На МР-ангиограммах, выполненных в режиме 3D TOF, визуализируются мозговые, внутренние сонные, позвоночные, основная артерии и их интракраниальные ветви. Со стороны сосудов головного мозга определяется снижение сигнала от позвоночной артерии справа. Сосуды симметричны, диаметр остальных не изменен, артериовенозные анастомозы отсутствуют. Венозные синусы свободно проходимы.
Заключение. МР-признаки дисциркуляторной энцефалопатии. Умеренная смешанная заместительная гидроцефалия.
Диагноз: СХА, начальная стадия.
От операции пока воздерживается.
Таким образом, СХА — малоизвестное, хотя и достаточно часто встречающееся заболевание, этиология которого не полностью ясна. Основные клинические проявления синдрома включают развитие атаксии, апраксии, явления деменции и нарушения мочеиспускания, вплоть до недержания мочи. Диагностика основывается на данных клинической симптоматики, люмбальной пункции с пробным извлечением жидкости и нейровизуализации. Основным способом лечения является наложение вентрикулоперитонеального шунта для отведения ЦСЖ. Своевременное проведение хирургического вмешательства дает хорошие результаты, улучшает качество жизни пациентов.
Синдром Морганьи-Адамса-Стокса
Синдром Морганьи-Адамса-Стокса - это комплекс симптомов, обусловленных резким снижением сердечного выброса и ишемией головного мозга у больных, страдающих выраженными нарушениями ритма. Проявляется в виде приступов синкопе, судорог, фибрилляции желудочков, асистолии. Диагноз устанавливается по наличию характерной клинической картины, изменениям на электрокардиограмме, результатам суточного мониторирования. Заболевание дифференцируют с эпилепсией, истерическим припадком. Лечение состоит из реанимационных мероприятий в момент развития симптоматики и последующего терапевтического восстановления нормальной работы сердца.
МКБ-10
Общие сведения
Впервые синдром Морганьи-Адамса-Стокса был описан итальянским анатомом и врачом Д. Морганьи в 1761 году. В период с 1791 по 1878 г заболевание изучалось ирландскими кардиологами Р. Адамсом и В. Стоксом. С учетом вклада всех специалистов синдром был назван их именами. Патология распространена среди пациентов, страдающих сердечными болезнями, в первую очередь - блокадами внутрисердечной проводимости и синдромом слабости синусового узла. Чаще диагностируется у людей старше 45-55 лет, мужчины составляют около 60% от общего числа больных. Максимальное количество случаев регистрируется в развитых странах, жители которых склонны к гиподинамии и подвержены воздействию кардиотоксических веществ. В государствах «третьего мира» синдром встречается сравнительно редко.
Причины
К развитию болезни приводят врожденные органические изменения в строении проводящей системы, а также нарушения, возникающие под влиянием внешних патогенетических факторов. К их числу относят передозировку антиаритмическими средствами (новокаинамид, амиодарон), профессиональную интоксикацию хлорорганическими соединениями (винилхлорид, четыреххлористый углерод), дистрофические и ишемические изменения миокарда, затрагивающие крупные узлы системы автоматизма (синатриальный, атриовентрикулярный). Кроме того, синдром может формироваться в результате возрастной дегенерации АВ-центра. Непосредственно приступ имеет следующие причины:
- Блокадыпроводимости. Наиболее распространенная этиологическая форма. Развивается при переходе неполной АВ-блокады в полную. При этом возникает диссоциация между предсердиями и желудочками. Первые сокращаются под действием импульсов, исходящих от СА-узла, вторые возбуждаются АВ-центром или эктопическими очагами. Также провоцирующим фактором может послужить частичная атриовентрикулярная блокада на фоне синусового ритма и увеличенного показателя сердечных сокращений.
- Нарушения ритма. Приступ выявляется при чрезмерном подъеме или уменьшении ЧСС. Обычно синкопе отмечается у пациентов с частотой сердечных сокращений более 200 или менее 30 ударов в минуту. При наличии диффузных поражений церебрального сосудистого аппарата потеря сознания наблюдается уже при пульсе 40-45 уд/мин. Патология также может потенцироваться фибрилляцией предсердий, особенно - возникшей впервые. Постоянные формы аритмии редко приводят к появлению симптоматики МАС.
- Утратасократительнойфункции. Происходит при вентрикулярной фибрилляции. Мышечные волокна миокарда сокращаются некоординированно, по отдельности, с очень высокой частотой. Это делает выброс крови невозможным, ведет к остановке кровообращения и развитию клинической смерти. Может встречаться при нарушениях электролитного баланса, иметь идеопатическую природу (на фоне полного здоровья), являться результатом воздействия физических факторов.
Патогенез
В основе лежит резкое сокращение сердечного выброса, которое становится причиной замедления кровотока, недостаточного снабжения органов и тканей кровью, кислородом, питательными веществами. Первоначально от гипоксии страдают нервные структуры, в том числе головной мозг. Работа ЦНС нарушается, происходит потеря сознания. Чуть позже возникают судорожные мышечные сокращения, свидетельствующие о выраженном кислородном голодании тканей. Длительные приступы, особенно обусловленные фибрилляцией желудочков, могут привести к постгипоксической энцефалопатии, полиорганной недостаточности. При сохранении минимального кровотока (блокады, аритмии) заболевание протекает легче. Приступы в большинстве случаев не приводят к отсроченным последствиям.
Классификация
Патогенетическая систематизация, учитывающая причины и механизмы формирования приступа, используется при плановом лечении и выборе мер профилактики. При оказании экстренной помощи синдром Морганьи-Адамса-Стокса удобнее классифицировать по виду нарушения коронарного ритма, поскольку это позволяет быстро определить оптимальную тактику лечения. Различают следующие виды патологии:
- Адинамическийтип. Наблюдается при отказе синоатриального узла, блокадах III и II степени, когда частота сокращений желудочков снижается до 20-25. Включает асистолию - остановку сердца, возникающую при резком и полном нарушении проводимости внутрисердечного импульса. До момента подключения альтернативных эктопических зон проходит достаточно много времени, что становится причиной прекращения кровообращения.
- Тахиаритмическийтип. Определяется при увеличении ЧСС до 200 в минуту и выше. Выявляется при синусовой тахикардии, трепетании, вентрикулярном мерцании, пароксизмальных суправентрикулярных ТК, фибрилляции предсердий с проведением импульса на желудочки по обходным путям при синдроме Вольфа-Паркинсона-Уайта.
- Смешанныйтип. Моменты предсердной или желудочковой тахикардии чередуются с эпизодами асистолии. Приступ развивается при быстром уменьшении ЧСС с высоких показателей до брадикардии или временной остановки сердца. Эта форма является наиболее сложной для диагностики и прогностически неблагоприятной.
Симптомы
Классический припадок характеризуется быстрым развитием и определенной последовательностью изменений. В течение 3-5 секунд с момента возникновения аритмии или блокады у пациента формируется предобморочное состояние. Внезапно появляется головокружение, головная боль, дискоординация, дезориентация, бледность. На коже выступает обильный холодный пот. При пальпаторной оценке пульса обнаруживается резко выраженная тахикардия, брадикардия или неровный ритм.
Второй этап длится 10-20 секунд. Больной теряет сознание. Снижается артериальное давление, мышечный тонус. Визуально определяется акроцианоз, развиваются мелкие клонические судороги. При фибрилляции желудочков отмечается симптом Геринга - своеобразное жужжание в области мечевидного отростка грудины. Через 20-40 секунд судороги усиливаются, приобретают эпилептовидную форму, происходит непроизвольное мочеиспускание, дефекация. Если ритм не восстанавливается через 1-5 минут, наблюдается клиническая смерть с исчезновением пульса, дыхания, роговичных рефлексов. Зрачок расширен, АД не определяется, кожа бледная, мраморного оттенка.
Возможно абортивное течение приступа с редукцией симптоматики в течение очень короткого временного промежутка. Кора мозга не успевает подвергнуться выраженной гипоксии. Основными симптомами, наблюдаемыми при этом варианте патологии, являются головокружение, слабость, преходящее нарушение зрения, помрачение сознания. Проявления исчезают за несколько секунд без медицинского вмешательства. Подобные разновидности МАС крайне сложно диагностировать, поскольку аналогичная симптоматика выявляется при множестве других состояний, в т. ч. при цереброваскулярной болезни.
Осложнения
Синдром приводит к ряду осложнений, основным из которых является клиническая смерть. Длительная остановка кровотока - фактор отмирания части клеток коры головного мозга. После успешной реанимации это становится причиной энцефалопатии, соматических нарушений, снижения умственных способностей больного. В число осложнений можно включить изменения психоэмоционального фона пациента, постоянно испытывающего страх перед наступлением нового криза, что негативно отражается на качестве жизни, продуктивности работы и отдыха. Во время утраты сознания и падения на землю больной может получить травмы, которые также относят к патологическим состояниям, ассоциированным со СМАС.
Диагностика
Первичную диагностику осуществляют сотрудники СМП, прибывшие на вызов. Окончательный диагноз устанавливает кардиолог, основываясь на результатах электрокардиографии и холтеровского мониторирования. Дифференциальную диагностику проводят с эпилептическим припадком, истерией. Отличительной особенностью истинной эпилепсии является смена тонических судорог клоническими, гиперемия лица, предшествующая аура. При истерическом происхождении патологии утраты сознания не происходит, присутствует синусовый сердечный ритм. Признаками болезни МАС считаются наличие аритмии того или иного характера, стремительное развитие клинической картины. В процессе диагностического поиска используют следующие методы:
- Физикальноеобследование. Отмечаются типичные симптомы, состояние развивается быстро (в течение нескольких десятков секунд). В анамнезе присутствуют заболевания кардиологического профиля, непосредственно перед приступом возможен эпизод психоэмоционального возбуждения, переживаний. АД резко снижено или не измеряется, сердечный ритм неровный. Тип нарушения можно определить только по результатам ЭКГ.
- Инструментальноеобследование. Основной метод аппаратной диагностики - электрокардиография. В момент появления симптомов на пленке регистрируются отрицательные расширенные зубцы «T» в отведениях V4-V2. Возможно присутствие деформированных желудочковых комплексов. При блокадах наблюдается диссоциация «P» с «QRS», косонисходящая депрессия сегмента «ST». Фибрилляция проявляется отсутствием нормальной активности на ЭКГ, появлением мелкой или крупной волнистой линии. С помощью суточного мониторирования удается обнаружить преходящую блокаду, на фоне которой наступает приступ.
- Лабораторноеобследование. Проводится с целью определения причин болезни и ее последствий. После эпизода клинической смерти выявляется изменение уровня pH в кислую сторону, дефицит электролитов, присутствие в крови миоглобина. При коронарных заболеваниях возможен рост кардиоспецифических маркеров: тропонина, КФК МВ.
Неотложная помощь
Помощь при синдроме МАС включает непосредственное купирование приступа и профилактику рецидивов. При развивающемся припадке спасательные мероприятия производит присутствующий медицинский работник независимо от его профиля и специализации. Осуществляются комбинированные реанимационные мероприятия. Лечение включает:
- Прекращениеприпадка. Применяется тот же алгоритм, что при остановке сердца. Рекомендовано проведение прекардиального удара, непрямого массажа, при отсутствии дыхания - ИВЛ методом рот в рот или с использованием соответствующей аппаратуры. При ФЖ производится электрическая дефибрилляция. Внутривенно вливается адреналин, атропин, хлористый кальций, инотропные средства. При тахиаритмии показаны антиаритмические препараты: амиодарон, новокаинамид.
- Предотвращениеприпадка. Если приступы обусловлены пароксизмами ТА, пациенту требуются препараты для стабилизации работы миокарда и выравнивания сердечного ритма. При блокадах медикаментозная терапия неэффективна, необходима имплантация асинхронного или деманд-кардиостимулятора. При реципрокной тахикардии возможно оперативное разрушение одного из проводящих путей АВ-узла.
Прогноз и профилактика
Прогноз благоприятный при быстром купировании приступа и при его абортивном варианте. Нормализация сердечного ритма и кровоснабжения головного мозга в течение 1 минуты с момента формирования клинической картины не сопровождается отсроченными последствиями. Длительный период асистолии или фибрилляции желудочков снижает вероятность благополучного восстановления коронарного ритма и повышает риск ишемического поражения головного мозга. Специфические меры профилактики не разработаны. Общие рекомендации по предотвращению кардиологических болезней включают отказ от курения и алкоголя, исключение гиподинамии, занятия спортом, соблюдение принципов здорового питания. При появлении первых признаков нарушений в работе сердца следует обратиться к врачу для обследования и лечения.
1. Приступ Морганьи-Адамса-Стокса при синдроме слабости синусового узла/ Батьянов И.С., Бухарова Г.В. - 2001.
Синдром Адамса-Кершнера (Adams-Kershner) - синонимы, авторы, клиника
Синдром Адамса-Оливера — это состояние, включающее сочетание дефектов черепа и определённого типа дефекта конечности.
Описание Синдром Адамса-Оливера — это генетическое состояние, характеризующееся аплазией, чаще всего волосистой части головы и черепа, и поперечными дефектами конечности. Также сообщалось о врождённом пороке сердца у лиц с этим заболеванием. Точная причина состояния не совсем понятна.
Содержание статьи:
Генетический профиль
Сообщалось о семейных и несемейных случаях синдрома Адамса-Оливера. Большинство генетических случаев были унаследованы аутосомно-доминантным способом, но также сообщалось аутосомно-рецессивное и спорадическое наследование. Разница в представлении в доминантной и рецессивной форме не документирована.
Аутосомно-доминантное наследование означает, что для возникновения заболевания требуется только одна аномальная копия гена. Для лиц с копией гена риск передачи его своему потомству один на два или на 50%.
Аутосомно-рецессивное наследование означает, что два дефектных экземпляра гена должны быть унаследованы, по одному от каждого родителя, чтобы болезнь проявлялась. Лица с одной генной мутацией являются носителями этого расстройства. Лица, которые являются носителями рецессивного типа синдрома Адамса-Оливера, не имеют никаких симптомов (бессимптомные) и не знают, что они являются носителями, если у них не было ребёнка с синдромом.
Тестирование не доступно, так как местоположение гена в настоящее время неизвестно. Вероятность того, что каждый член пары будет носителем мутации в том же гене, выше у людей, которые связаны генетически (называются кровными родственниками).
Существует два-три шанса, что здоровый родственник пострадавшего ребёнка является носителем. Спорадические проявления могут быть вызваны доминантным геном с переменной экспрессией (ни у кого в семье нет симптомов, но на самом деле некоторые из них являются носителями генов), новая (доминантная) мутация, возникающая во время формирования эмбриона, где ни один из родителей не является носителем как генетических, так и негенетических причин одного и того же синдрома.
Были постулированы различные механизмы для объяснения того, как происходит синдром Адамса-Оливера. К ним относятся:
- травма,
- сжатие матки,
- амниотическая полоса (состояние, возникающее из нитей мембраны амниона, вызывающее ампутацию частей плода),
- нарушение сосудов (блокирование притока крови к развивающейся части или части плода)
- и большой сгусток крови в плаценте, который блокирует некоторые важные кровеносные сосуды и прерывает кровоснабжение развивающихся структур.
Недавно было высказано предположение о том, что синдром Адамса-Оливера возникает в результате аномалий в небольших структурах сосудов, которые встречаются очень рано при формировании эмбриона. Аномалия сосудов может быть результатом генетического дефекта, вызывающего снижение стабильности эмбриональных кровеносных сосудов при наличии определённых сил.
Демография
Признаки и симптомы синдрома Адамса-Оливера
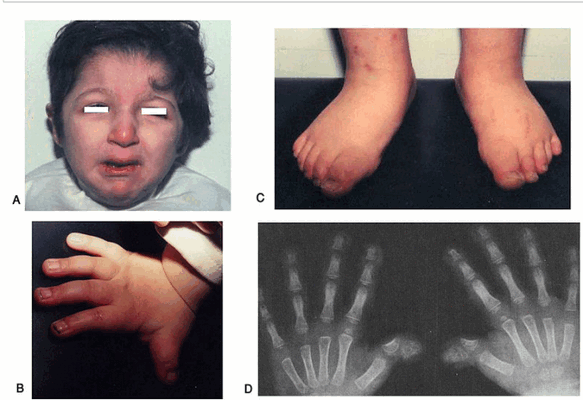
Дефекты конечностей являются наиболее распространённым явлением синдрома Адамса-Оливера, затрагивая около 84% пациентов.
Тип дефекта конечности обычно асимметричный (не одинаковый с обеих сторон), с тенденцией к вовлечению обеих сторон тела (двусторонний), чаще нижних конечностей, чем верхних конечностей. Существует широкий диапазон серьёзности дефектов конечности, от чего-то минимального, такого как маленький или отсутствующий палец или ноготь (гипоплазия ногтей), до более сильного отсутствия рук, ног или нижних конечностей.
Сообщается о других более умеренных дефектах конечности, которые включают в себя перевязку (синдактилию) кожи (кожная синдактилия) или кости (костная синдактилия) пальцев рук или ног, деформацию когтевой руки и брахидактилию (укороченные пальцы).
Реже дефекты черепа видны без дефектов кожи головы и могут быть ошибочно приняты за увеличенное мягкое пятно (родничок). Сообщается, что врождённые пороки сердца встречаются у 13-20% пациентов с синдромом Адамса-Оливера. У этих пациентов сообщалось о многих различных типах сосудистых (включая кровеносные сосуды) и клапанных (с участием сердечных клапанов) проблем. Другие клинические признаки, включают в себя:
- небольшой рост,
- почечные пороки развития,
- расщепление нёба,
- маленькие глаза (микрофтальмия),
- лишние соски,
- неопущение яичек,
- поражения кожи
- неврологические аномалии,
- в нескольких случаях присутствует умственная отсталость.
Диагностика
Чтобы определить, имеет ли пациент синдром Адамса-Оливера, соответствующие члены семьи должны быть осмотрены для подтверждения состояния. Когда аплазия обнаружена при рождении, следует оценить плаценту. Физическое обследование пострадавшего младенца включает оценку других связанных структур, особенно зубов, волос и других областей кожи, ногтей и центральной нервной системы.
Как только эта оценка будет завершена и будет определён или опровергнут конкретный диагноз синдрома Адамса-Оливера, можно обеспечить генетическое консультирование. Возможна пренатальная диагностика ультразвуком дефектов конечности и, возможно, некоторые другие аномалии, но клиническое подтверждение диагноза происходит после рождения. Так как ген (или гены), вызывающие синдром, не были изолированы, пренатальные диагностические процедуры, такие как амниоцентез или отбор хорионического ворсинок, не нужны.
Лечение
Лечение для синдрома отличается для каждого человека и адаптировано к конкретным симптомам. Если присутствует несоответствие длины ног, можно носить корректирующие туфли, которые увеличивают подошву для неповреждённой ноги, чтобы предотвратить сколиоз и трудности с ходьбой.
Иногда рекомендуются ортопедические устройства, такие как протезы. Пациентов следует направлять к врачу, специализирующемуся на лечении пациентов с дефектами конечности на раннем этапе жизни. Может потребоваться хирургическое вмешательство для врождённых дефектов и пересадки кожи для дефектов кожи головы (около 30% пациентов нуждаются в пересадке кожи). Около 30% пациентов в одном исследовании страдали серьёзным кровотечением от дефекта кожи головы. В таких случаях может потребоваться лечение, такое как трансфузия или антибиотикотерапия. Соответствующие специальные образовательные услуги необходимы для лиц с умственной отсталостью.
Нормотензивная гидроцефалия: симптомы, диагностика и лечение

Нормотензивная гидроцефалия (также — синдром Хакима-Адамса) является заболеванием, которое клинически проявляется неупорядоченной походкой, нарушением когнитивной функции и удержания.
Эти симптомы, в особенности у пациентов старшего возраста, являются довольно распространённым явлением и их не всегда легко связать с подозрением на наличие гидроцефалии.
Этиопатогенез и патофизиология
В анамнезе некоторых пациентов с подозрением на синдром Хакима-Адамса можно найти ушиб, черепно-мозговую травму, субарахноидальное кровоизлияние или воспаление центральной нервной системы, которые на протяжении многих лет вели к расширению желудочковой системы и впоследствии - к декомпенсации и клиническим проявлениям.
У большинства пациентов, однако, присутствует идиопатическое заболевание, где чёткая причинно-следственная связь не наблюдается.
С патофизиологической точки зрения признаётся гидродинамический концепт повышенного давления ликворовых пульсаций в желудочках без увеличения внутрижелудочкового давления, что приводит к образованию гидроцефалии.
Причин нарушения дренажа спинномозговой жидкости может быть много. Оно может быть связано с проявлением врождённых дефектов, менингитом, опухолями, кровоизлияниями в мозг, являться последствием инсульта или травмы головы, а также развиваться в виду возраста.
Разновидности протекания нарушения
Существует два типа течения синдрома Хакима-Адамса — в острой и хронической формах.
Функциональная классификация нормотензивной гидроцефалии:
- обструктивная;
- коммуникативная;
- гиперсекреторная;
- гипорезорптивная.
С точки зрения динамики (важно для лечения)

Клиническая картина
Симптомы, указывающие на развитие нормотензивной гидроцефалии:
- нарушение ходьбы (фронтальная апраксия): замедление ходьбы, сокращение шага, снижение высоты шага;
- нарушение когнитивных способностей, напоминающие признаки слабоумия - апатия, снижение концентрации, потеря интереса, проблемы с памятью, психомоторное замедление в целом;
- нарушение континенции - сначала прерывистые нарушение удержания мочи вплоть до недержания, включая недержание кала.
Симптомы могут быть по-разному выражены. Чаще всего сначала проявляются нарушение ходьбы, которая предшествует когнитивным расстройствам, в последнюю очередь развивается нарушение континенции.
Дополнительные симптомы, которые могут присутствовать (головные боли, шум в ушах, психоаффективные расстройства) не являются типичными, но не исключают диагноза НГ. Как правило, они характерны для пациентов старше 60 лет, ввиду чего эти клинические проявления могут быть отнесены к старению и их возникновение может быть недооценено лечащим врачом.
Методы постановки диагноза
Для диагностики заболевания используют следующие способы исследования.
КТ головного мозга
При подозрении на заболевание необходимо провести КТ головного мозга, которая засвидетельствует расширение желудочковой системы.
Следует исключить другие патологические изменения, приводящие к гидроцефалии (опухоли, врождённые аномалии, пороки развития сосудов) и возможную гидроцефалию «ex vacuo», при которой расширение желудочковой системы развивается от атрофии мозга и клинические проявления могут быть сходными.
На основании результатов КТ и клинических подозрений на нормотензивную гидроцефалию пациента осматривает невролог, который при помощи структурно более отвечающего требованиям МРТ и клинической оценки проводит дифференциальный диагноз (другие виды деменции и расстройства походки - дегенеративные, интоксикационные, метаболические, инфекционные), и после исключения других причин направляет больного в соответствующее отделение для дальнейшего клинического исследования, проверки ходьбы и проведения когнитивных тестов.
В случае подозрения на заболевание выполняется ликвородинамическое обследование.
Спинномозговая пункция
Самый простой тест - спинномозговая пункция, в ходе котором игла вводится в поясничный отдел позвоночного канала в поясничной области и измеряется внутричерепное давление (потенциальное исключение внутричерепной гипертензии), после чего отбирается 30-50 мл спинно-мозговой жидкости (СМЖ). Чувствительность теста составляет около 50-60%.
Люмбальный дренаж
При отрицательных приведённых выше тестах, но при высоком клиническом подозрении можно выполнить люмбальный дренаж с чувствительностью до 90%.
Непрерывный дренаж спинномозговой жидкости 10 мл/ч в течение 3-х дней имитирует введение ВП шунта и приводит к клиническому улучшению.
Возможности терапии
Основным методом лечения синдрома Хакима-Адамса является введение вентрикулоперитонеального (ВП) шунта. Операция занимает около часа, и в её ходе обрабатывается боковой желудочек, который соединён через трубку с клапаном, ведённым подкожной тканью брюшной полости.
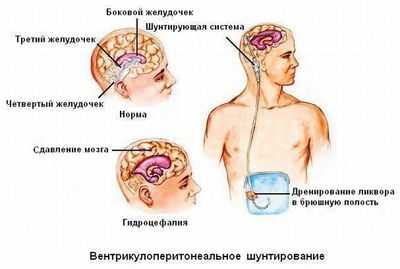
Есть и альтернативы - вентрикулоатриальный и люмбоперитонеальный шунт. В последние годы также выполняется вентикулостромия, при которой эндоскопически фенестрируется 3 желудочек.
Контроль КТ мозга осуществляется на 2-3 день после операции в целях избегания возможных осложнений. К ним относятся кровотечение, неправильное положение шунта и др. В долгосрочной перспективе может дойти к инфекции, что может привести к нарушению закрытия шунта и необходимости повторного лечения.
К снижению числа инфекций приводит пропитка антибиотиками вводимых шунтов. Риск инфекции в течение первого года составил всего до 5%.
При правильной индикации происходит относительно быстрое и значительное улучшение расстройств походки. Улучшение расстройств континенции является постепенным, и менее положительно на него реагируют пациенты с выраженным значительным ухудшением когнитивных способностей.
После эффективного лечения, тем не менее, необходимо регулярный клинический мониторинг с помощью КТ (1 раз в год).
Осложнения и прогноз
Частота осложнений после операций у больных с НГ в литературе различается. Смертность не превышает 2% и, по мнению некоторых авторов, в большей мере связана с сопутствующими заболеваниями, чем с имплантацией шунта.
Образование субдуральных нарушений приводится в диапазоне 2-17%. Процент этих осложнений значительно снижается с использованием современных программируемых клапанов.
Риск инфекционных осложнений относительно низок, и не превышает 5-6%. Другие осложнения могут заключаться в неисправности самого шунта, образовании гематом после пункции желудочков и даже развитие эпилепсии.
Важную роль играет длительность ухудшения ходьбы перед операцией, и пациенты, у которых расстройство походки продолжалось менее 1-го года, имеют наилучший прогноз.
Положительный эффект приведённых выше методов лечения заболевания составляет 70-80% пациентов. В течение нескольких лет происходит снижение эффекта шунта, также необходимо регулярно снижать давление перепуска.
Чтобы не стать жертвой заболевания, человек должен быть осведомлён о мерах предосторожности. Необходимо всегда покрывать голову при рисковой работе, использовать шлемы при езде на мотоцикле, вовремя обнаруживать и лечить инфекционные и воспалительные заболевания, не допускать переохлаждения головы.
Нормотензивная гидроцефалия. Синдром Хакима-Адамса

Заболевание было описано впервые в 1965 г. S. Hakim и R. D. Adams. [1, 3]. Является вариантом открытой гидроцефалии. Для данного заболевания характерно медленное расширение желудочковой системы при нормальном давлении цереброспинальной жидкости. Это приводит к развитию триады симптомов (триада Хакима-Адамса). К ним относятся нарушение ходьбы, деменция и недержание мочи. [1].
Эпидемиология. В литературе встречаются разные данные на этот счет. Заболевание, как правило, поражает людей пожилого возраста. Диагноз ставится 0. 41% населения старше 65 лет, 0. 4-6% пациентам с деменцией и 15% с нарушением ходьбы [1]. D. Jaraj и соавт. (2014) отмечали, что нормотензивная гидроцефалия встречается в 0, 2% случаев в возрасте 70 - 79 лет и в 5, 9% - 80 лет и старше. Однако авторы придерживаются мнения, что реальные показатели значительно выше. Примечательно, что описаны случаи диагностирования данного заболевания даже в детском возрасте. Распространенность между мужчинами и женщинами одинаковая. [2].
Этиология. Выделяют первичную и вторичную нормотензивную гидроцефалию. В первом случае выявить причин развития заболевания не удается. Такие пациенты составляют от половины [1, 2] до одной трети [3] случаев. Вторичная гидроцефалия является следствием субарахноидального кровоизлияния (30J, менингита (15%), черепно-мозговой травмы (10%), нейрохирургических операций. [3].
Патогенез. В основе заболевания формирование дисбаланса между секрецией и резорбцией цереброспинальной жидкости, нарушение ликвородинамики. В результате увеличивается объем ликворного пространства, уменьшается объем мозговой ткани [2]. Это приводит к необратимым ишемическим и дегенеративным белого и серого вещества головного мозга. При этом характерно преобладание лобного характера неврологических нарушений. Считается, что это связано с преимущественным расширением передних рогов боковых желудочков, что приводит к сдавлению глубинных отделов лобных долей, передних отделов мозолистого тела, двигательных путей, связывающих кору с нижними конечностями, разобщением базальных ядер с фронтальной корой, дисфункцией лобных долей, нарушением сенсомоторной интеграции. [1]. В некоторых случаях наблюдается снижение поступления крови в головной мозг. [2].
Клиническая картина. В клинике заболевания характерно постепенное развитие классической триады симптомов (нарушение ходьбы, деменция и недержание мочи) в течение нескольких месяцев или лет [1, 3]. Однако после черепно-мозговой травмы или субарахноидального кровоизлияния возможно появление симптомов в первые дни и недели. [1].
Первыми признаками заболевания обычно появляются нарушения ходьбы. Походка замедляется, затем становится неустойчивой, возможны падения. Далее проявляется апраксия ходьбы (неуверенность при стоянии и ходьбе, затрудненное начало движения). При этом лежа и сидя пациент легко имитирует движения ходьбы, а в вертикальном положении это мгновенно утрачивается. Сила в конечностях не нарушена. [1]. Может выявляться постуральный тремор, акинетико-ригидный синдром (феномен «застывания»). Это сближает заболевание с ригидной формой болезни Паркинсона (данный диагноз часто выставляется первоначально). Однако при обследовании ригидность мышц не выявляется. Иногда встречается псевдобульбарный синдром. [2].
Для когнитивных нарушений характерен лобно-подкорковый характер, развиваются обычно на фоне уже существующих моторных проявлений. [1]. Проявляются выпадения кратковременной и долговременной памяти, дезориентированность во времени. Пациенты с трудом излагают свой анамнез. Возникают проблемы в планировании, сосредоточении, абстрактном мышлении, нарушения семантической памяти. Оскудевает эмоциональная сторона, появляются апатия, благодушие. Нередки явления агнозии (нарушение различных видов восприятия: зрительного, слухового, тактильного). Замедляется скорость психических процессов и психомоторных реакций. Степень нарушений когнитивных функций различна. [2].
Нарушение функции тазовых органов на ранних этапах выявляется при активном расспросе - учащенное мочеиспускание и никтурия. Далее появляются императивные позывы и затем недержание мочи. При прогрессировании когнитивных нарушений пациенты теряют критику к данной проблеме и относятся к ней индифферентно. [1, 2].
Диагностика. Диагноз ставится на основании триады симптомов с расширением желудочковой системы при нормальном уровне внутричерепного давления.
1) наличие полной или неполной триады Хакима-Адамса (нарушение походки, когнитивные нарушения, нарушение контроля над функцией тазовых органов, в первую очередь - мочеиспусканием)
2) наличие характерной рентгенологической картины НТГ, которая включает сочетание следующих признаков:
- расширение желудочков [головного] мозга: индекс Эванса более 0, 3 (30%) и расширение височных рогов боковых желудочков более 2 мм (для справки: желудочково-полушарный индекс Эванса представляет собой отношение расстояния между наиболее отдаленными точками передних рогов боковых желудочков к наибольшему внутреннему диаметру черепа)
- диспропорциональное расширение субарахноидальных пространств (DESH-симптом): расширение цистерн основания черепа, боковых щелей мозга в сочетании с компрессией межполушарной щели и парасагиттальных субарахноидальных пространств в теменной области [2].
В постановке диагноза также используется проведение люмбальной пункции с опредением давления ликвора. Давление ликвора остается нормальным при данном заболевании [1, 2].
Кроме того, проводится ТАП-ТЕСТ (TAP-TEST). Его также называют тестом Миллера-Фишера, люмбальным или спинальным тестом. Проводится следующим образом: производится люмбальная пункция с выведение 30-50 мл ликвора. До и после выполнения пробы проводят видеозапись походки. Проба считается положительной, если после эвакуации ликвора отмечается значимое улучшение походки или других симптомов. Положительная проба подтверждает диагноз нормотензивной гидроцефалии. Пока окончательно не решено, когда и как оценивать результаты этого теста. В основном оценка производится через 1 сутки. При нормотензивной гидроцефалии после такой процедуры походка и когнитивные функции временно улучшаются. Однако K. Kang и соавт. (2013) описали пациента, у которого через 1 сутки отклика не было, но улучшение наступило через 7 суток. [2]. Степень улучшения состояния пациента после проведения теста совпадает с эффектом шунтирующей операции. Считается даже кратковременное уменьшение проявлений хоть одного из симптомов заболевания благоприятным прогностическим признаком. [1].
Лечение. Основным методом лечения данных пациентов являются шунтирующие операции с наложением вентрикулоперитонеального, вентрикулоатриально или люмбоперитонеального шунта. Положительные результаты наблюдаются у 60% [4] -66% [1] -75% [2] пациентов. Прогноз оперативного вмешательства ухудшается по мере прогрессирования заболевания. [1]. Предпочтительно использование клапанно-регулируемым системам с анти-сифонным устройством и системам с программируемым клапаном переменного давления с максимально возможным конструктивно малым шагом изменения открывающего давления. [2].
Важно отметить, что по данным некоторых авторов даже при отсутствии положительного эффекта от тап-теста, шунтирующие операции имели значительный положительный результат. [4, 5, 6].
В послеоперационном периоде необходимо комплексное реабилитационное лечение.
Читайте также:
- Техника амбулаторных операций на глотке, гортани. Аденотомия, тонзиллотомия
- Психоз у ребенка: клиника, диагностика
- Программирование медицинской помощи. Индивидуальные пакеты помощи
- Рентгенограмма, КТ, МРТ, УЗИ при отрыве ужерживателя сгибателей стопы
- Глазодвигательные нервы: ядра, зоны иннервации, нарушения функции