Синдром Барттера (Bartter) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 01.02.2026
Синдром Барттера - наследственный дефект почечных канальцев, вызывающий снижение уровня калия, хлоридов, что порождает метаболический алкалоз.
Представляет собой набор тесно связанных расстройств. Они различаются в зависимости от возраста начала появления симптомов, величины выделения калия (K), простагландинов в моче и степени экскреции кальция в моче.
Описание
Различают три клинических фенотипа:
- Неонатальный (или антенатальный), называемый синдром гиперпростагландина Е.
- Классический синдром Барттера
- Синдром Гительмана
Существует множество пациентов, у которых есть другой вариант, который, по их мнению, может быть вариантом Bartter’s, который еще не идентифицирован.
Характерны комбинации классического Бартера и Гительмана. Из проведенных исследований выяснилось, что многие люди, имеющие Classic Bartter первоначально, меняются по мере взросления и начинают испытывать проблемы с магнием, что более характерно для Gitelman’s.
Это озадачивает многих нефрологов и исследователей. Пациенты переходят с классического Барттера на Гительмана. Никто не знает, почему это происходит.
Классический синдром Барттера
Синдром Бартера был впервые обнаружен в 1962 году Фредериком Барттером. Бартер описал его у двух афроамериканских пациентов: 5-летнего мальчика и 25-летнего мужчины с длительной историей медленного роста, слабости и усталости.
При больших рационах натрия оба пациента имели нормальное кровяное давление и высокую экскрецию альдостерона в моче, что приводило к метаболическому алкалозу. Пациенты проявляют множество клинических симптомов:
- Глубокая гипокалиемия (очень низкие уровни калия в сыворотке)
- Увеличение экскреции мочи калием (К) и простагландинами.
- Нормальное кровяное давление, несмотря на повышенный уровень ренина плазмы и альдостерона. (Высокий ренин и альдостерон вызывает гипертензию у нормального здорового человека. У Бартерра Ренин и Альдостерон повышен, но нет гипертонии)
- Гипохлормический (низкий уровень сывороточного хлорида) метаболический алкалоз.
- Относительное сосудистое сопротивление прессорным эффектам экзогенного ангиотензина II
- Гиперплазия юкстагломерулярного аппарата
Таблица. Корреляции генотипа-фенотипа
| Генетический тип | Дефектный ген | Клинический тип |
| Bartter тип I | NKCC2 | неонатальный |
| Bartter тип II | ROMK | неонатальный |
| тип III | CLCNKB | классический |
| Bartter тип IV | BSND | Неонатальный с глухотой |
| тип IVb | CLCNKB иCLCNKA | Неонатальный с глухотой |
| Bartter тип V | CaSR | классический |
Первоначально считался сосудистым заболеванием. В 1970-х годах, когда были обнаружены простагландины, стало понятно, что пациенты с синдромом Бартера перепроизводили их. При лечении ингибитором уровень альдостерона возвращается к норме.
Впоследствии экспериментальный дефицит калия вызывал продукцию простагландина и многие симптомы синдрома Бартера. Проблема оказалась не внутрисосудистой, а почечной трубчатой.
Распространенность
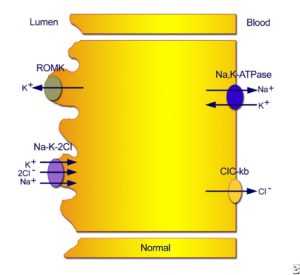
Нормальные транспортные механизмы в толстой восходящей ветке петли Генле
Имеются как семейные (Унаследованные), так и спорадические формы синдромов Барттера и Гительмана. Распространенность расстройства точно не известна, но в одном исследовании приведена оценка 1,2 на миллион.
Хотя многие случаи кажутся спорадическими, Барттер хорошо описан у братьев и сестер. Схема передачи предполагает аутосомно-рецессивный способ наследования.
Нет гендерного предпочтения и расовой предрасположенности.
Классический синдром Барттера обычно диагностируется в детстве или подростковом возрасте. Неонатальный может быть заподозрен до рождения или диагностируется сразу после рождения.
В классической форме симптомы начинаются у новорожденных или у детей возраста 2 лет или младше. Синдром Гительмана у детей часто не диагностируется до подросткового или раннего взрослого возраста.
Симптомы
Признаки и симптомы антенатальных синдромов Барттера, известные как синдромы Бартера 1 и 2 и синдромы Бартера 4а и 4b, можно наблюдать до рождения (антенатальный период).
Аномальная функция почек в матке может привести к чрезмерному производству мочи и аномальному накоплению околоплодной жидкости вокруг развивающегося плода (полигидрамниоза).
Рождение часто преждевременно. В новорожденный период пораженные дети испытывают чрезмерное мочеиспускание (полиурия) и опасные для жизни эпизоды лихорадки и обезвоживания. Также могут возникать рвота и диарея.
Внешний вид
Некоторые затронутые младенцы имеют характерные черты лица - треугольной формы, видный лоб, большие глаза, большие, заостренные уши и «надутое» выражение из-за поникших углов рта.
В некоторых случаях эти отличительные черты могут отсутствовать или настолько мягкие, что остаются незамеченными. Пострадавшие младенцы не развиваются, не набирают вес. Окончательный рост взрослых ниже, чем можно ожидать.
У некоторых людей, которые испытывают значительный дисбаланс электролита, развиваются нерегулярные сердечные сокращения (сердечные аритмии). Если не лечить, сердечные аритмии прогрессируют, могут вызвать внезапную остановку сердца.
- Усталость;
- Полиурия (повышенное мочеиспускание);
- Полидипсия (повышенная жажда);
- Nocturia (Просыпаются ночью, чтобы помочиться);
- Общая слабость;
- Повышенная потребность в соли;
- Дегидратация;
- Умственная путаница;
- Рвота;
- Мышечная слабость;
- Мышечные спазмы;
- Тетания;
- Неспособность развиваться;
- Низкий рост (если не лечить).
Диагностика
Диагностика синдрома Бартера основана на выявлении характерных симптомов, детальной истории болезни, тщательной клинической оценке и различных специализированных тестах.
Клинические испытания
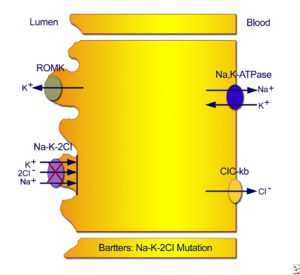
Тип I неонатальный синдром Барттера. Мутации в хлорангидриде натрия хлорид / калий хлорид приводит к дефектной реабсорбции натрия, хлорида и калия
Лабораторные тесты, которые используются для диагностики этих расстройств.
Например, анализы крови для определения уровней электролита, в частности уровня магния, ренина, альдостерона, анализы мочи для определения присутствия простагландина E2, электролитов, включая повышенные уровни натрия, калия.
Антенатальные подтипы могут быть диагностированы до рождения (пренатально), когда полигидрамниоз обнаруживается без наличия связанных с ним врожденных пороков развития.
Повышенные уровни хлорида и альдостерона обнаруживаются в околоплодной жидкости.
Молекулярно-генетическое тестирование подтверждает диагноз. Оно обнаруживает мутации в определенных генах, но доступно только в специализированных лабораториях.
Лабораторные результаты
- Низкий уровень калия в сыворотке;
- Низко-нормальные уровни магния;
- Увеличение ренина;
- Повышенный альдостерон;
- Метаболический алкалоз;
- Повышенная экскреция простагландина е2;
- Нормальная выдержка кальция в моче;
- Выращивание Mg с нормальным высвобождением;
- Нормально низкий уровень сыворотки Mg;
- Нормальное - низкое кровяное давление;
- Повышенная экскреция калия в моче;
- Повышенный плазменный ангиотензин II;
- Нефрокальциноз;
- Tetany, мышечные спазмы, знак Chvostek и знак Trousseau могут быть замечены в hypokalemia, hypocalcemia, и hypomagnesemia пациентов. В старой литературе иногда значился рахит.
Исследование
В 1997 году Мадригал описал тип синдрома в Коста-Рике у шестнадцати из двадцати пациентов со «своеобразной фацией, отличающейся треугольной формой лица, большими глазами, выступающими ушами».
- Еще восемь имели сенсоневральную потерю слуха, определяемую по аудиографии.
В дополнение к этим биохимическим нарушениям, небольшое количество пациентов развило прогрессирующую почечную недостаточность из-за тяжелого тубуло-интерстициального нефрита.
Неясно, является ли потеря функции почек у этих пациентов прямым следствием их первичного дефекта или вторично к хронической гипокалиемии.
Дифференциальный диагноз
Синдром псевдо-Бартера является общим термином, который относится к определенным состояниям, которые вызывают те же симптомы и признаки синдрома Барттера, но в которых нет почечной трубчатой дисфункции.
При использование определенных диуретиков часто наблюдается рвота, булимия, синдром циклической рвоты, злоупотребление слабительными.
У некоторых людей с кистозным фиброзом могут наблюдаться симптомы, подобные тем, которые наблюдаются при синдроме Барттера, особенно у маленьких детей с кистозным фиброзом, которые имеют специфические факторы риска (например, тяжелое респираторное или панкреатическое заболевание, желудочно-кишечные расстройства).
Что вызывает
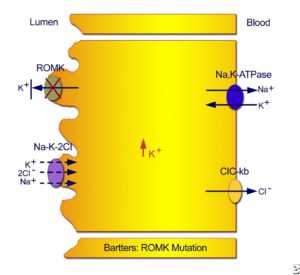
Тип II неонатальный синдром Барттера. Мутации в гене РОМК приводят к невозможности рециркулировать калий из клетки обратно в трубчатый просвет, что приводит к ингибированию хлорангидрида хлорида натрия / хлорида калия
Синдром Бартера вызван изменениями (мутациями) в одном из нескольких разных генов. Большинство подтипов наследуются аутосомно-рецессивным способом.
Наиболее приемлемым объяснением синдрома Классического Бартера является первичный дефект в транспорте Cl в TAL. Это часть одного из канальцев в почках, где электролиты переходят в кровоток.
Рецессивные генетические расстройства возникают, когда индивидуум наследует две копии аномального гена для одного и того же признака, по одному от каждого родителя.
Если человек наследует один нормальный ген и один ген заболевания, он будет носителем заболевания, но без симптомов.
Риск иметь ребенка, который является носителем, таким как родители, составляет 50% при каждой беременности. Шанс на получение ребенком нормальных генов у обоих родителей составляет 25%. Риск одинаковый для мужчин и женщин.
Большинство генов, участвующих в расстройстве, продуцируют (кодируют) белки, необходимые для правильного здоровья и функции почек.
Лечение
Лекарства от синдрома Бартера нет. Лечение направлено на конкретные симптомы, требует пожизненного приема определенных добавок и препаратов.
- Добавки для хлористого калия;
- Магниевые добавки;
- Спиронолактон;
- Amilioride;
- Triamterene;
- Индометацин;
- Каптоприл;
- Гормон роста.
Педиатры, общие терапевты, специалисты по почкам (нефрологи), социальные работники и другие специалисты области здравоохранения нуждаются в систематическом и всестороннем планировании лечения ребенка. Генетическая консультация полезна пострадавшим людям и их семьям. Также необходима психосоциальная поддержка всей семьи.
Основа лечения - восстановление правильного баланса жидкостей и электролитов в организме. Например, добавление хлорида калия, чтобы помочь устранить дисбаланс электролита. Добавка хлорида калия предпочтительнее добавок соли из-за соответствующих хлоридных недостатков.
Некоторым детям с тяжелыми, опасными для жизни заболеваниями петли (антенатальные синдромы Барттера) нужна замена соли и воды через центральный венозный катетер. Поскольку агрессивная замена жидкости ухудшает полиурию, может потребоваться лечение препаратом, таким как индометацин, который предотвращает производство простагландина 2.
Индометацин - нестероидный противовоспалительный препарат (NSAID). Он снижает уровень простагландина в организме, тем самым уменьшая избыточное производство мочи, потребность добавок калия. Индометацин обычно эффективен и хорошо переносится.
Некоторые затронутые индивидуумы могут принимать лекарства, известные как калийсберегающие диуретики, такие как спиронолактон или амилорид.
Эти препараты увеличивают выделение воды в моче, но сохраняют калий, предотвращающий гипокалиемию. Они не всегда оказываются эффективными при лечении гипокалиемии, и могут ухудшить потерю соли в организме (почечная соль).
Ингибиторы
Препараты, которые ингибируют или блокируют ренин-альдостерон-ангиотензиновую систему (ингибиторы РААС), используются в дополнение к другим методам терапии (вспомогательная терапия). Ингибиторы RAAS включают антагонисты альдостерона, блокаторы рецепторов ангиотензина II, ингибиторы ангиотензинпревращающего фермента (ACE).
Эти препараты предотвращают секрецию альдостерона из надпочечников, противодействуют действию ренина на почки, тем самым уменьшая потерю калия.
Их использование необходимо контролировать, потому что понижают артериальное давление, которое может быть снижено у людей с синдромами Бартера, влияют на функцию почек, сердечно-сосудистой системы.
- Гормональная терапия успешна для лечения замедления роста.
- В некоторых случаях для лечения мышечных спазмов или тетаний требуется добавление кальция или магния.
- Требуется достаточное потребление соли и воды. Затронутые люди имеют большой аппетит к соли из-за соляной тяги. Пострадавшим может быть предложено употреблять в пищу продукты с высоким содержанием калия.
- Кохлеарные имплантаты используются для лечения глухоты, связанной с синдромами Бартера типа 4А и 4В.
В стрессовых ситуациях электролиты крови быстро меняются, требуют быстрого внутривенного лечения. Стрессовые ситуации могут включать хирургические процедуры, травму и наличие другого типа заболевания или инфекции (интеркуррентное заболевание).
Прогноз
Синдром Барттера и Гительмана - аутосомно-рецессивные расстройства, и ни одно из них не излечимо.
Степень инвалидности зависит от тяжести дисфункции рецептора, но прогноз во многих случаях хорош, при этом пациенты способны вести нормальную жизнь.
Имеющаяся ограниченная прогностическая информация свидетельствует о том, что ранняя диагностика и соответствующее лечение детей грудного, раннего возраста с синдромом Классического Бартера улучшает рост и нейроинтеллектуальное развитие.
С другой стороны, устойчивая гипокалиемия, гиперревинемия вызывают прогрессирующий тубулоинтерстициальный нефрит, приводящий к почечной недостаточности.
При раннем лечении дисбалансов электролитов прогноз для пациентов с синдромом Классического Бартера является хорошим.
- При лечении уровень ренина в плазме, уровень альдостерона нормализуются. Терапия улучшает клиническое состояние, позволяет увеличить рост.
- Возраст костей обычно подходит для хронологического возраста, а пубертатное и интеллектуальное развитие являются нормальными с лечением.
Установлена эффективность долгосрочного использования ингибиторов простагландинов синтетазы. Иногда наблюдается рецидив гипокалиемии, которой можно управлять, регулируя дозу индометацина или добавку калия. Болезнь не рецидивирует у пациентов с пересаженной почкой.
Заболеваемость и смертность
Значительная заболеваемость и смертность возникают, если синдром Барттера не лечится. Долгосрочный прогноз остается спорным из-за медленного прогрессирования хронической почечной недостаточности и интерстициального фиброза.
- Сенсоневральная глухота обусловлена дефектами субъединицы бартина в каналах ClC-Ka и CIC-Kb.
- Нефрокальциноз часто ассоциируется с гиперкальциурией.
- Почечная недостаточность. Почечная недостаточность довольно необычна. При обзоре 63 пациентов 5 развили прогрессирующую почечную болезнь, требующую диализа или трансплантации.
- Иногда развивается обратимая острая почечная недостаточность от рабдомиолиза из-за гипокалиемии.
- Низкий рост. Почти у всех наблюдается замедление роста.
Дополнительные осложнения
- Сердечная аритмия и внезапная смерть - результат электролитного дисбаланса
- Задержка развития - Общая для людей без лечения
- Значительное снижение минеральной плотности костей - у пациентов с неонатальной или классической формой.
Обучение пациентов
Пациенты и их родители должны понимать, что не существует никакого лечения мутаций, вызывающих различные формы синдрома Барттера. Это хроническое состояние требует регулярного приема лекарств, как это предписано, что часто бывает трудно для детей и подростков.
Пациенты должны знать о возможных неблагоприятных последствиях медицинской терапии, особенно раздражения желудочно-кишечного тракта, кровотечения.
Люди, как правило, истощаются, если ограничены натрием и водой. Должна быть обеспечена адекватная замена жидкости и электролита, особенно в жаркую погоду, во время физических упражнений.
Пациентам следует избегать интенсивных упражнений из-за опасности обезвоживания и функциональных нарушений сердечной деятельности, вторичных по отношению к дисбалансу калия.
Что касается диеты, необходимо знать, какие продукты имеют высокое содержание калия.
БАРТТЕРА СИНДРОМ
БАРТТЕРА СИНДРОМ (описан американским эндокринологом F. C. Bartter, 1914-1983; синоним - гипокалиемический алкалоз) - редкое наследственное заболевание из группы тубулопатий, характеризующееся повышенным образованием почечных простагландинов и нарушением реабсорбции хлора в толстом сегменте восходящей части петли Генле. Потеря хлорида натрия обусловливает активацию ренин-ангиотензиновой системы с развитием гипокалиемии и метаболического алкалоза. Заболевание проявляется в детском возрасте, отмечаются задержка роста, умственная отсталость, полиурия, дегидратация, судороги, мышечная гипотония, эпизоды лихорадки и диареи. Отеков нет, уровень артериального давления остается нормальным. При обследовании выявляют повышенное выведение калия, кальция, простагландинов с мочой, гипокалиемию, метаболический алкалоз, повышенную активность ренина плазмы и альдостерона, гиперплазию юкстагломерулярного аппарата в биоптате почки. Тип наследования - аутосомно-рецессивный. Лечение симптоматическое: средства, блокирующие продукцию почечных простагландинов, ингибиторы ангиотензин-превращающего фермента, препараты калия, антагонисты альдостерона и др.
F. C. Bartter, P. Pronove, J. R. Gill Jr, R. C. MacCardle. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. American Journal of Medicine, New York, 1962; 33: 811-828.
Энциклопедический словарь по психологии и педагогике . 2013 .
Полезное
Смотреть что такое "БАРТТЕРА СИНДРОМ" в других словарях:
ШВАРЦА - БАРТТЕРА СИНДРОМ — (по именам американских врачей W. B. Schwartz, род. в 1922, и F. C. Bartter, 1914-1983; синоним - синдром неадекватной секреции антидиуретического гормона) - связан с избыточной секрецией антидиуретического гормона (вазопрессина), что приводит к… … Энциклопедический словарь по психологии и педагогике
Синдром Барттера — Схема сосудистых коммуникаций почечных канальцев (петля Генле) … Википедия
Синдром де Тони — Дебре Фанкони МКБ 10 E72.072.0 МКБ 9 270.0270.0 DiseasesDB … Википедия
Синдром Клайнфельтера — Кариотип … Википедия
Синдром гиперкортицизма — Синдром Кушинга МКБ 10 E24.24. МКБ 9 255.0255.0 MedlinePlus … Википедия
Синдром Конна — Синдром Конна … Википедия
Синдром Нельсона — Синдром Нельсона … Википедия
Синдром Пархона — МКБ 10 E22.222.2 МКБ 9 253.6253.6 DiseasesDB … Википедия
Синдром поликистозных яичников — Поликистозный яичник: ультразвуковое изображение МКБ 10 E … Википедия
СИНДРОМ БАРТТЕРА
СИНДРОМ БАРТТЕРА мед.
Синдром Барттера — наследственное заболевание (см. также Приложение 2. Наследственные болезни: картированные фенотипы) с выраженным снижением ОЦК из-за потери электролитов с почками, сочетающееся с низким АД, гипокалиемическим алкалозом, гиперкальциурией и нормальным содержанием магния в сыворотке. Последние две особенности отличают пациентов с этим синдромом от пациентов с синдромом Гительмана, у которых, в дополнение к гипокалиемическому алкалозу и потере солей, обнаруживают гипокальциурию и гипомагниемию. Пациенты тяжело больны с рождения, и при длительном клиническом течении часто развивается нефрокальциноз, ведущий к почечной недостаточности.
Клиническая картина
• Отсутствие артериальной гипер-тёнзии — важный дифференциально-диагностический признак
• Отставание в физическом и умственном развитии
• Мышечная слабость и мышечные судороги.
Диагностика
• Эритроцитоз
• Снижение агрегации тромбоцитов
• Значительное повышение содержания ренина и альдостерона в плазме
• Гиперхлорурия (гипохлоремия)
• Гипокалиемия
• Гипонатриемия
• Гипо-магниемия
• Гиперкальциемия и гиперкальциурия
• Гиперфосфатемия
• Повышение экскреции с мочой простагландинов и калликреина.
Лечение
• Диета с богатым содержанием калия и хлоридов
• Заместительная терапия препаратами калия
• Спиронолактон
• Каптоприл
• Пропранолол (анаприлин)
• Индометацин.
Синонимы
• Алкалоз гипокалиемический с гиперкальциурией
• Алкалоз гипокалиемический
?26.8 ?ругие формы гиперальдостеронизма МШ
• 241200 Синдром Барттера
• 263800 Синдром Гительмана
Литература
Bartter FC et al: Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. Am. J. Med. 33: 811-828, 1962; Simon DB et al: Mutations in the gene CLCNKB cause Bartter's syndrome type III. Nature Genet. 17: 171-178, 1997
Синдром Барттера - это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.
Общие сведения
Синдром Барттера в клинической урологии представляет собой редкую генную мутацию - дефект петли Генле, наследуемую по аутосомно-рецессивному типу и проявляющуюся, как правило, уже в детском возрасте. Неспособность почечных нефронов задерживать калий приводит к хронической потере его с мочой и уменьшению объема циркулирующей крови при нормальном или пониженном АД. В зависимости от вида пораженных генов различают: неонатальный синдром Барттера 1 и 2 типов, классический синдром Барттера, синдром Гительмана. Также встречается приобретенный синдром псевдо-Барттера, характеризующийся сходными проявлениями, но не сопровождающийся патологией почечных канальцев.
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике - сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
Синдро́м Ба́рттера (но́рмотензи́вный ги́перальдостерони́зм, неонатальный синдром Барттера) — форма гиперальдостеронизма с гиперплазией юкстагломерулярного аппарата почек и резистентностью к сосудосуживающему действию ангиотензина II, обусловленой внешними (вторичными) нарушениями передачи сигнала ангиотензина II [1] .
Содержание
История
Синдром назван в честь доктора Фредерика Барттера, который вместе с доктором Pacita Pronove, первым описал его в 1960 году и нескольких пациентов в 1962 году [1] [2] [3] [4] [4] .
Характеристика синдромов Барттера и Гительмана
Синдромы характеризуется гипокалиемией, нормальным или пониженным артериальным давлением, и наличием гипохлоремического метаболического алкалоза [5] .
Псевдо-синдром Барттера
Псевдо-синдром Барттера (Pseudo-Bartter’s syndrome) — симптомокомплекс, иммитирующий проявления аналогичные синдрому Барттера, однако не сопровождается характерными генетическими дефектами и проявляется кистозным фиброзом [6] , а также развивается на фоне злоупотребления слабительными средствами [7]
Этиология
Тип наследования аутосомно-рецессивный, андротропизм [1] . Синдром вызывают мутации генов, кодирующих белки, обеспечивающие транспорт ионов в почечных канальцах [8] .
Синдром может быть разделён на различные подтипы в зависимости от мутировавшего гена [9] :
| Название | Тип синдрома Барттера | Мутация гена, кодирующего | Дефект |
| Неонатальный синдром Барттера | тип 1 | NKCC2 | Na-K-2Cl symporter |
| Неонатальный синдром Барттера | тип 2 | ROMK | thick ascending limb K + каналы |
| Классический синдром Барттера | тип 3 | CLCNKB | Cl - каналы |
| Синдром Барттера с нейросенсорной тугоухостью | тип 4 | BSND [10] | Cl - channel accessory subunit |
| Синдром Барттера, ассоциированный с аутосомно-доминантной гипокальциемией | тип 5 | CASR [11] | activating mutation of the calcium-sensing receptor |
| Синдром Гительмана | - | SLC12A3 (NCCT) | Sodium-chloride symporter |
Патогенез
Первичное звено — потеря способности почек задерживать калий. Возможные причины: нарушение функции почечных канальцев, нарушение реабсорбции хлоридов в петле Генле (приводит к увеличению поступления натрия и воды в дистальные отделы канальца, где происходит секреция калия), гипомагниемия. Потеря калия ведёт к многочисленным метаболическим и гормональным расстройствам: нарушению транспорта веществ через клеточные мембраны, усилению продукции сосудорасширяющих простагландинов, снижение секреции альдостерона, повышение продукции простагландинов в почках [1] .
Клиническая картина
Первые признаки заболевания обычно проявляются в течение первого года жизни. Клинические проявления синдрома [1] :
- , , , (жажда), ,
- гипернатриемия, ,
- гиперальдостеронурия, .
Пренатальная диагностика — синдром Барттера можно заподозрить при многоводии [12] .
Чувствительность к ангиотензину II повышается или полностью восстанавливается после нормализации объёма внеклеточной жидкости и при лечении ингибиторами простагландинсинтетазы (даже на фоне сохраняющейся гипокалиемии) [1] .
Пациентам рекомендуют диету, обогащённую калием и приём спиронолактона для уменьшения потери калия с мочой. [13]
См. также
Примечания
Ссылки
Синдром Пархона (неадекватной продукции АДГ) • Синдром Сотоса (церебральный гигантизм) • Синдром Пехкранца — Бабинского — Фрелиха (адипозо-генитальная дистрофия) • Синдром Каллмана • Синдром Килина (анорексия)
Читайте также: