Синдром буллезного эпидермолиза - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 28.01.2026
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
Филиал "Вешняковский" Московского научно-практического центра дерматовенерологии и косметологии Департамента здравоохранения Москвы
ФГУ "Федеральный научный центр трансплантологии и искусственных органов им. В.И. Шумакова" Минздрава России, Москва
Особенности течения и диагностики приобретенного буллезного эпидермолиза (клинико-иммунопатологическое наблюдение)
Журнал: Клиническая дерматология и венерология. 2013;11(2): 92‑97
Махнева Н.В., Нефедова Е.Д., Померанцев О.Н., Белецкая Л.В. Особенности течения и диагностики приобретенного буллезного эпидермолиза (клинико-иммунопатологическое наблюдение). Клиническая дерматология и венерология. 2013;11(2):92‑97.
Makhneva NV, Nefedova ED, Pomerantsev ON, Beletskaia LV. The course and diagnosis of acquired epidermolysis bullosa (a clinical and immunopathological observation). Klinicheskaya Dermatologiya i Venerologiya. 2013;11(2):92‑97. (In Russ.).
Представлен клинический случай приобретенного буллезного эпидермолиза смешанного генеза с участием IgG/IgA-аутоантител, имитирующего клиническую картину вульгарной пузырчатки, торпидно протекающего на фоне иммуносупрессивной терапии. Рассматривая вопросы этиопатогенеза, авторы подчеркивают важную роль иммунофлюоресценции в диагностике и дифференциальной диагностике аутоиммунных буллезных дерматозов.
Приобретенный буллезный эпидермолиз (ПБЭ) — редкое аутоиммунное буллезное заболевание кожи и слизистых оболочек, встречающееся в 0,17—0,26% случаев на 1 млн человек в Северной и Западной Европе [1, 2]. Несмотря на то что не обнаружено преимуществ выявления этого дерматоза в зависимости от расовой и половой принадлежности, отмечено его широкое распространение среди корейской популяции [3]. В Московской области за 1992—2006 гг. диагностировано всего 9 случаев ПБЭ при клинико-лабораторном обследовании 267 больных, страдающих аутоиммунными буллезными дерматозами [4].
Причина возникновения ПБЭ до конца не известна. Однако наличие в сыворотке крови у больных ПБЭ аутоантител, направленных к компонентам базальной мембраны кожи человека и животных (к детерминантам коллагена VII типа), свидетельствует о том, что буллезный дерматоз имеет аутоиммунный механизм развития [5—8]. Антитела больных ПБЭ относятся в основном к IgG [9, 10].
Основным клиническим признаком ПБЭ является возникновение спонтанных или травмоиндуцированных пузырей в зрелом возрасте (в 40—50 лет) с отсутствием семейной предрасположенности и исключением других буллезных дерматозов [11—13]. Как правило, в патологический процесс вовлекаются кожа и слизистая оболочка. При классическом варианте ПБЭ основным клиническим отличительным признаком является первичное возникновение пузырей и эрозивных дефектов на месте травм без или с минимальным проявлением воспалительного процесса. Это связано с физической слабостью кожного покрова, особенно в выступающих местах и местах сдавления (локтевых и коленных сгибов, сводов стоп) с образованием рубцов, милиа и/или эритемы [13—15]. Возможно развитие ониходистрофии и рубцовой алопеции [16, 17].
При воспалительном варианте ПБЭ пузыри, как правило, возникают на воспаленном участке кожи без травматизации на открытых ее участках. Клиническая картина отличается полиморфизмом и напоминает буллезный или рубцующийся пемфигоид [14, 18, 19].
Мы наблюдали за пациентом с ПЭБ смешанного генеза с участием IgG/IgA-аутоантител, имитирующего клиническую картину вульгарной пузырчатки, торпидно протекающего на фоне иммуносупрессивной терапии.
Обследован больной Ш., 54 лет. Болен около 6 лет (с 2006 г.). Впервые появились буллезные элементы на слизистой оболочке полости рта, коже волосистой части головы и верхней трети спины. Начало заболевания пациент связывает с избыточной инсоляцией и нервным перенапряжением. Клинически диагностирована вульгарная пузырчатка.
В Городской клинической больнице №14 им. В.Г. Короленко проведено лечение преднизолоном в максимальной начальной дозе (100 мг/сут внутрь) с постепенным ее снижением. Больной выписан с клинически положительным эффектом (эпителизацией эрозивных дефектов и отсутствием свежих буллезных элементов при дозе преднизолона 35 мг/сут). В течение 3 мес после выписки доза преднизолона была снижена до поддерживающей (10 мг/сут внутрь), на фоне которой периодически возникали единичные пузыри на коже конечностей, которые эпителизировались после применения спрея оксикорт. Патологический процесс постепенно прогрессировал и в 2009 г. пациент с обострением процесса госпитализирован в кожную клинику МИА им. И.М. Сеченова для обследования и коррекции терапии. При обследовании данных, свидетельствующих за паранеопластический процесс, не выявлено. Иммуноморфологическая картина интактного участка кожи свидетельствовала в пользу буллезного пемфигоида. Обнаружена фиксация IgG, IgA и С3-компонента комплемента в зоне базальной мембраны эпидермиса. Однако клинический диагноз «вульгарная пузырчатка» не был снят. Проведено лечение: метипред 20 таблеток в сутки (80 мг) в течение 21 дня. В связи с торпидным течением и отсутствием положительной динамики от проводимой терапии назначена комбинированная терапия преднизолоном (80 мг/сут) и азатиоприном (150 мг/сут). Однако патологический процесс прогрессировал.
К лечению были добавлены дипроспан (2,0 мл внутримышечно 1 раз в 5 дней, №3) и плазмаферез (8 сеансов). На фоне указанной терапии достигнут клинически положительный эффект (отсутствие свежих высыпаний, полная эпителизация эрозий). Пациент выписан на дозе преднизолона 25 мг/сут внутрь с рекомендациями о ее постепенном снижении. С 2010 г. пациент находится на поддерживающей терапии преднизолона (10 мг/сут внутрь). В течение последующих 1,5 лет у пациента периодически возникают единичные буллезные элементы и эрозивные дефекты на коже конечностей в местах травмирования, которые эпителизировались на фоне топических глюкокортикостероидов. С декабря 2011 г. отмечено обострение патологического процесса. Больной госпитализирован с диагнозом «вульгарная пузырчатка, обострение».
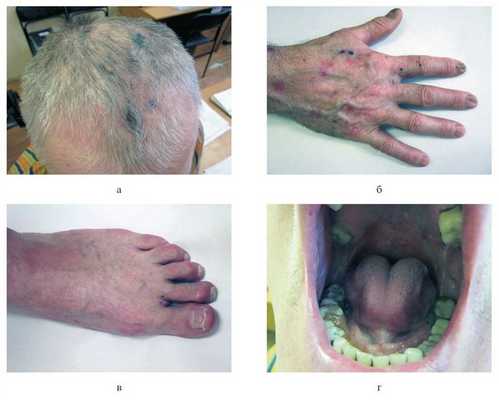
01.06.12 больной поступил в 1-е стационарное отделение филиала «Вешняковский» Московского научно-практического центра дерматовенерологии и косметологии Департамента здравоохранения Москвы (история болезни №12/246). Общее состояние удовлетворительное. Ведущие жалобы: высыпания на коже волосистой части головы, верхних и нижних конечностей, слизистой оболочке полости рта, жжение в местах высыпаний, боли при глотании и в эпигастрии. Пациент отмечал появление пузырей и эрозий непосредственно в местах ушиба или сдавления. При осмотре: процесс носит распространенный, подостровоспалительный характер с локализацией на коже волосистой части головы, туловища, верхних и нижних конечностей, слизистой оболочке полости рта. На коже волосистой части головы в проекции теменных зон процесс представлен эритематозно-сквамозными очагами и единичными эрозиями, расположенными на воспалительном фоне, покрытыми серозно-геморрагическими корочками размером до 1,5 см в диаметре (рис. 1, a). Рисунок 1. Клинические проявления ПБЭ. a — лобно-теменная зона: единичные эрозивные дефекты (до 0,5 см) на эритематозном фоне с серозно-геморрагическими корочками, эритематозносквамозные очаги; б — кожа правой кисти: эрозии (до 0,7 см в диаметре) с серозным отделяемым, серозно-геморрагическими корочками; поствоспалительные гиперпигментированные и эритематозные пятна (диаметром до 1,5 см), милиумы (до 0,3 см в диаметре); ногтевые пластины дистрофичны (тусклые, желтоватые, истонченные, продольно исчерченные); в — левая стопа: ногтевые пластины тусклые, деформированы, с продольной исчерченностью, множественными лейконихиями и крошащимися краями; г — полость рта. На коже туловища, конечностей — единичные эрозии до 0,7 см в диаметре, частично покрытые серозно-геморрагическими корочками с серозным отделяемым; на месте бывших высыпаний — гиперпигментированные пятна диаметром до 1,5 см. Симптом Никольского отрицательный. На коже кистей — милиумы (см. рис. 1, б). Ногтевые пластины тусклые, деформированы, продольно исчерчены, крошатся (см. рис. 1, в). На слизистой оболочке полости рта (щек, мягкого и твердого неба) патологический процесс представлен эрозиями с ярко-розовым дном размером до 0,7 см в диаметре, окаймленными по периферии обрывками эпителия беловатого цвета (см. рис. 1, г).
Из анамнеза: острые респираторные вирусные инфекции до 5—6 раз в год; в 2011 г. — операция по поводу правосторонней паховой грыжи.
Для исключения неопластического процесса как одного из факторов, провоцирующих буллезный дерматоз, пациенту проведено комплексное обследование. Диагностированы ишемическая болезнь сердца; стенокардия напряжения I—II функционального класса; гипертоническая болезнь II ст.; язвенная болезнь двенадцатиперстной кишки, рубцовая деформация луковицы двенадцатиперстной кишки; хронический панкреатит в ремиссии; мочекаменная болезнь, конкременты почек, киста синуса левой почки; хронический субатрофический фарингит. По данным эзофагогастродуоденоскопии, рубцовая деформация луковицы двенадцатиперстной кишки, поверхностный гастрит и косвенные признаки хронического панкреатита. В клиническом анализе крови — лейкоцитоз (до 15,3×10 9 /л). Данных за онкопатологию не обнаружено.
При цитологическом исследовании мазков-отпечатков со дна эрозий слизистой оболочки полости рта и кожи акантолитические клетки (клетки Тцанка) не обнаружены. При микроскопическом исследовании гладкой кожи и ногтевых пластин стоп грибы не выявлены.
На основании анамнеза, клинико-морфологической картины, особенности течения болезни и результатов ранее проведенного иммуногистохимического исследования пациенту выставлен диагноз ПБЭ.
Проведено лечение преднизолоном (60 мг/сут внутрь) с постепенным снижением дозы (до 50 мг/сут), на фоне которого на слизистой оболочке полости рта сохранялись эрозивные дефекты. Пациент переведен на комбинированную терапию глюкокортикостероидами: преднизолон (30 мг/сут) и метипред (16 мг/сут) внутрь. Лечение без эффекта. К терапии присоединен сандиммун-неорал в дозе 400 мг/сут ежедневно с последующим ее снижением до 300 мг/сут. На фоне приема сандиммун-неорала появились боли в эпигастральной и поясничной областях, и препарат был постепенно отменен. Комбинированный прием преднизолона (30 мг/сут внутрь) и метипреда (16 мг/сут внутрь) продолжен.
Из сопутствующей терапии: гемодез (200,0 мл внутривенно капельно через сутки, №3), трентал (5,0 мл внутривенно капельно с 200 мл физиологического раствора, №10), актовегин (5,0 мл внутримышечно ежедневно, №10), эссенциале (5,0 мл внутривенно струйно, №15), витамины В1 и В6 (по 1,0 мл внутримышечно, чередование), фолиевая кислота 1 мг (по 1 таблетке 2 раза в сутки), аспаркам (по 1 таблетке 3 раза в сутки), форкан 150 мг (1 капсула в неделю), омепразол 20 мг (1 капсула 2 раза в сутки), фосфалюгель (1 пакетик 3 раза в сутки), кальций D3-никомед 0,5 мг (по 1 таблетке 2 раза в сутки), эналаприл (10 мг 2 раза в сутки), индапамид (1,5 мг утром), гипотиазид (25 мг утром), нитроглицерин (0,5 мг при болях в области сердца). Местная терапия: на эрозивные дефекты на коже волосистой части головы, туловища — водный раствор метиленового синего, спрей оксикорт. Обработка слизистой оболочки полости рта тетраборатом натрия, полоскание полости рта отварами трав (шалфей, ромашка).
На фоне терапии эрозии на коже туловища, конечностей эпителизировались, на месте бывших высыпаний остались поствоспалительные пятна розово-красного цвета, милиумы на коже кистей, предплечий. На слизистой оболочке полости рта — эрозии в стадии эпителизации. Боли при глотании значительно уменьшились. Больной выписан с положительным клиническим эффектом на комбинированной терапии преднизолоном (30 мг/сут внутрь) и метипредом (16 мг/сут внутрь) под наблюдение дерматолога по месту жительства. Даны рекомендации.
ПБЭ — крайне редкая патология. Впервые ПБЭ описан во II половине XX века и получил свое название в связи с клиническим сходством с дистрофической формой врожденного буллезного эпидермолиза [20], который развивается у детей вследствие врожденного дефекта в гене, кодирующем коллаген VII типа (якорные фибриллы). Коллагены (фибриллярные и нефибриллярные) являются структурными элементами большинства форм соединительной ткани в организме человека и мутации в генах, кодирующих их, являются причиной развития разных заболеваний человека [21—25]. Это могут быть несовершенный остеогенез, некоторые формы остеопороза, хондродисплазия, сосудистые аневризмы, заболевание почек и буллезный эпидермолиз [26, 27]. В последнем случае дефектное связывание волокон коллагена VII типа с недостаточным количеством якорных фибрилл вызывает несостоятельность кожного покрова — его неустойчивость к механическим воздействиям с развитием травмоиндуцированных пузырей, которые могут располагаться как на спокойном, так и на минимально воспаленном фоне с образованием рубцов и милиа [13, 14, 16, 17]. Возможно развитие и ониходистрофии [11, 16, 17], что подтверждает и представленный нами клинический случай.
Ультраструктурное изучение кожи при ПБЭ позволило обнаружить аморфное состояние электронно-плотной полосы непосредственно под lamina densa с недостаточным количеством якорных фибрилл, которые крепят эпидермис к дерме, пересекая базальную мембрану [28—30]. Однако выявление в сыворотке крови у пациентов с данной патологией IgG-аутоантител к коллагену VII типа позволило предположить, что ПБЭ имеет аутоиммунный механизм развития, в котором якорные фибриллы скорее скомпрометированы аутоантителами, чем дефектами гена [6, 31]. Антитела не являются специфичными только для антигенов базальной мембраны эпидермиса. Они также взаимодействуют с антигенами базальных мембран слизистой оболочки полости рта, пищевода, влагалища [6, 12]. Ответственность аутоантител за проявления ПБЭ подтверждена экспериментально in vivo на новорожденных мышах [31—34]. Проведена пассивная передача аутоантител (очищенный IgG) от пациентов с ПБЭ новорожденным мышам. При этом у животных развивался процесс с гистологическими и иммунопатологическими изменениями, характерными для этого заболевания.
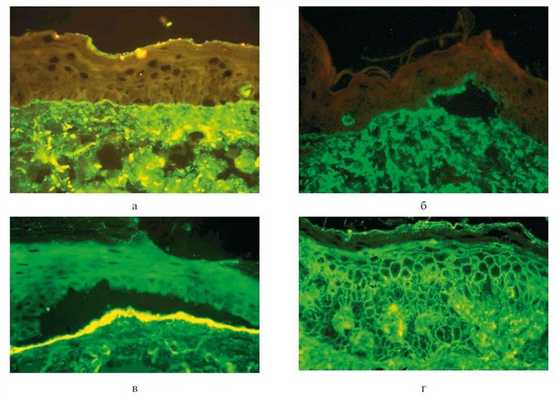
По мнению ряда авторов [35—37], в некоторых случаях патогенетическая роль при ПБЭ принадлежит циркулирующим аутоантителам класса A или одновременно IgA и IgG. Последнее подтверждает и описанный нами случай. С помощью методов меченых антител (прямой метод иммунофлюоресценции) в зоне базальной мембраны эпидермиса обнаружены связанные с тканями иммуноглобулины (IgA, IgG) и С3-компонент комплемента. Базальная мембрана эпидермиса является антигеном-мишенью для аутоантител буллезного пемфигоида и приобретенного буллезного эпидермолиза (рис. 2, a). Рисунок 2. Криостатные срезы кожи больных аутоиммунными буллезными дерматозами (для сравнительного контроля). Участки клинически непораженной кожи. Обработка меченой сывороткой против Ig G человека. Прямой метод иммунофлюоресценции (×400). a — буллезный пемфигоид: фиксация IgG в зоне базальной мембраны эпидермиса; б — в месте образовании пузыря фиксированный иммуноглобулин выявляется на покрышке в связи с фиксацией его в lamina lucida; в — фиксация IgG на дне подэпидермального пузыря (широкая щель) при ПБЭ; г — фиксация IgG в межклеточной субстанции всех слоев эпидермиса и на поверхности кожи при аутоиммунной пузырчатке. При этом фиксация аутоантител в lamina lucida и сохранение фиксированного иммуноглобулина на покрышке пузыря свидетельствуют о наличии буллезного пемфигоида (см. рис. 2, б), в то время как при ПБЭ фиксированный иммуноглобулин сохраняется на дне пузыря (см. рис. 2, в). При ПБЭ мишенью антител является lamina densa [38, 39].
В описанном нами случае ранее использованный метод меченых антител позволил исключить у пациента наличие аутоиммунной пузырчатки, для которой характерна картина фиксации IgG в межклеточной субстанции многослойного плоского эпителия (см. рис. 2, г) и заподозрить аутоиммунный буллезный дерматоз дермо-эпидермального соединения, в частности буллезный пемфигоид. Отсутствие подэпидермального микропузыря в криостатных срезах клинически интактного участка кожи пациента, к сожалению, не позволило иммуногистохимически уточнить мишень для фиксированных иммунных комплексов. Тем не менее тщательно собранный анамнез с указанием пациента на легкую ранимость кожного покрова, клинические проявления (рубцы, милиум, ониходистрофия), волнообразное течение болезни и торпидность патологического процесса к иммуносупрессивной терапии свидетельствовали в пользу ПБЭ.
Таким образом, ПБЭ — редкий буллезный дерматоз, имеющий специфические и неспецифические клинические проявления, лечение которого для врача представляет сложности. Это связано с волнообразным течением данного дерматоза, торпидного к иммуносупрессивной терапии. Неуклонное прогрессирование патологического процесса с вовлечением слизистых оболочек, угрожающего жизни пациента, требует от специалистов постановки точного диагноза с использованием молекулярно-биологических методов исследования, а от исследователей — изучения этиопатогенетических механизмов болезни с целью разработки более совершенных методов диагностики и лечения.
Синдром Лайелла ( Злокачественная пузырчатка , Острый эпидермальный некролиз , Токсический эпидермальный некролиз )
Синдром Лайелла — это тяжелое полиэтиологическое заболевание аллергической природы, характеризующееся острым нарушением общего состояния пациента, буллезным поражением всего кожного покрова и слизистых. Быстрое развитие обезвоживания, токсическое поражение почек и других внутренних органов, присоединение инфекционного процесса часто приводят к летальному исходу заболевания. Диагностика синдрома Лайелла включает объективное обследование пациента, постоянный мониторинг данных коагулограммы, клинических и биохимических анализов крови и мочи. Лечение синдрома Лайелла включает проведение неотложных мероприятий, методы экстракорпорального очищения крови, инфузионную терапию, введение больших доз преднизолона, антибиотикотерапию, коррекцию водно-солевых нарушений и др.
МКБ-10
Общие сведения
Синдром Лайелла (острый или токсический эпидермальный некролиз) относится к группе буллезных дерматитов. Свое основное название он получил в честь врача Лайелла, который в 1956 году впервые описал синдром как тяжелую форму токсикодермии. Клиническая картина синдрома Лайелла сходна с ожогом кожи II степени, в связи с чем заболевание называют ожоговым кожным синдромом. Еще одно распространенное название синдрома — злокачественная пузырчатка — обусловлено образованием на коже пузырей, подобных элементам пузырчатки.
Синдром Лайелла встречается в 0,3% случаев медикаментозных аллергий. После анафилактического шока он является самой тяжелой аллергической реакцией. Чаще всего синдром Лайелла наблюдается у людей молодого возраста и детей. Симптомы заболевания могут проявиться через пару часов или в течение недели после введения медикамента. По различным данным смертность при синдроме Лайелла составляет от 30% до 70%.

Причины
В зависимости от причины развития синдрома Лайелла современная дерматология выделяет 4 варианта заболевания.
- Первый представляет собой аллергическую реакцию на инфекционный процесс и чаще всего обусловлен золотистым стафилококком II группы. Как правило, он развивается у детей и отличается наиболее тяжелым течением.
- Второй — синдром Лайелла, который наблюдается в связи с применением лекарственных препаратов (сульфаниламидов, антибиотиков, противосудорожных лекарств, ацетилсалициловой кислоты, обезболивающих, противовоспалительных и противотуберкулезных средств). Наиболее часто развитие синдрома обусловлено одновременным приемом нескольких препаратов, одним из которых был сульфаниламид. В последние годы описаны случаи развития синдрома Лайелла на применение биологически активных добавок, витаминов, контрастных веществ для проведения рентгенографии и др.
- Третий вариант синдрома Лайелла составляют идеопатические случаи заболевания, причина возникновения которых остается невыясненной. Четвертый - синдром Лайелла, вызванный комбинированными причинами: инфекционными и лекарственными, развивается на фоне терапии инфекционного заболевания.
Патогенез
Большая роль в развитии синдрома Лайелла отводится генетически обусловленной предрасположенности организма к различным аллергическим реакциям. В анамнезе многих пациентов есть указания на аллергические заболевания: аллергический ринит, поллиноз, аллергический контактный дерматит, экзему, бронхиальную астму и др. У таких лиц из-за нарушения механизмов обезвреживания токсических продуктов обмена веществ происходит соединение введенного в организм лекарственного вещества с белком, который содержится в клетках эпидермиса. Это вновь образовавшееся вещество и является антигеном при синдроме Лайелла. Таким образом, иммунный ответ организма направлен не только на введенное лекарство, но и на кожу больного. Процесс напоминает реакцию отторжения трансплантата, в которой за трансплантат иммунная система принимает собственную кожу пациента.
В основе синдрома Лайелла лежит феномен Шварцмана-Санарелли — иммунологическая реакция, приводящая к нарушению регуляции распада белковых веществ и накоплению продуктов этого распада в организме. В результате происходит токсическое поражение органов и систем. Это нарушает работу обезвреживающих и выводящих токсины органов, что усугубляет интоксикацию, приводит к выраженным изменениям водно-солевого и электролитного баланса в организме. Данные процессы приводят к быстрому ухудшению состояния пациента при синдроме Лайелла и могут стать причиной летального исхода.
Симптомы
Синдром Лайелла начинается с внезапного и беспричинного повышения температуры тела до 39-40°С. За несколько часов на коже туловища, конечностей, лица, слизистой ротовой полости и гениталий появляются слегка отечные и болезненные эритематозные пятна различного размера. Они могут частично сливаться.
Через некоторое время (в среднем 12 часов) на участках внешне здоровой кожи начинает происходить отслаивание эпидермиса. При этом образуются тонкостенные вялые пузыри неправильной формы, величина которых варьирует от размеров лесного ореха до 10-15 см в диаметре. После вскрытия пузырей остаются большие эрозии, по периферии они покрыты обрывками покрышек пузырей. Эрозии окружены отечной и гиперемированной кожей. Они выделяют обильный серозно-кровянистый экссудат, что является причиной быстрого обезвоживания организма пациента.
При синдроме Лайелла отмечается характерный для пузырчатки симптом Никольского — отслаивание эпидермиса в ответ на незначительное поверхностное воздействие на кожу. В связи с отслойкой эпидермиса, на тех участках кожного покрова, которые подверглись сдавлению, трению или мацерации, эрозии формируются сразу, без образования пузырей.
Довольно быстро вся кожа пациента с синдромом Лайелла становится красной и резко болезненной при дотрагивании, ее внешний вид напоминает ожог кипятком II-III степени. Наблюдается характерный симптом «смоченного белья», когда кожа при прикосновении к ней легко сдвигается и сморщивается. В отдельных случаях синдрома Лайелла его основные проявления сопровождаются появлением мелкой петехиальной сыпи по всему телу больного. У детей заболевание обычно начинается с симптомов конъюнктивита и сочетается с инфекционным поражением кожи стафилококковой флорой.
Поражение слизистых при синдроме Лайелла проявляется образованием на них болезненных поверхностных дефектов, кровоточащих даже при незначительном травмировании. Процесс может затрагивать не только рот и губы, а и слизистую глаз, глотки, гортани, трахеи, бронхов, мочевого пузыря и уретры, желудка и кишечника.
Осложнения
Общее состояние пациентов с синдромом Лайелла прогрессивно ухудшается и за короткий период времени становится крайне тяжелым. Мучительная жажда, снижение потоотделения и продукции слюны являются признаками обезвоживания организма. Пациенты жалуются на выраженную головную боль, теряют ориентацию, становятся сонливыми. Наблюдается выпадение волос и ногтей. Обезвоживание приводит к сгущению крови и нарушению кровоснабжения внутренних органов. Наряду с токсическим поражением организма это приводит к нарушению работы печени, сердца, легких и почек. Развивается анурия и острая почечная недостаточность. Возможно присоединение вторичной инфекции.
Диагностика
Клинический анализ крови при синдроме Лайелла указывает на воспалительный процесс. Наблюдается повышение СОЭ, лейкоцитоз с появлением незрелых форм. Снижение или полное отсутствие эозинофилов в анализе крови является диагностическим признаком, позволяющим отличить синдром Лайелла от других аллергических состояний. Данные коагулограммы указывают на повышенную свертываемость крови. Анализ мочи и биохимический анализ крови позволяют выявить нарушения, происходящие со стороны почек, и осуществлять мониторинг состояния организма в процессе лечения.
Важной задачей является определение медикамента, который привел к развитию синдрома Лайелла, ведь его повторное применение в процессе лечения может быть губительным для пациента. Выявить провоцирующее вещество помогает проведение иммунологических тестов. На провоцирующий препарат указывает быстрое размножение иммунных клеток, возникающее в ответ на его введение в образец крови пациента.
Биопсия кожи и гистологическое изучение полученного образца у больного синдромом Лайелла обнаруживает полную гибель клеток поверхностного слоя эпидермиса. В более глубоких слоях наблюдается образование крупных пузырей, отечность и скопления иммунных клеток с наибольшей концентрацией в области кожных сосудов.
Синдром Лайелла дифференцируют с другими острыми дерматитами, сопровождающимися образованием пузырей: актиническим дерматитом, пузырчаткой, контактным дерматитом, синдромом Стивенса-Джонсона, буллезным эпидермолизом, герпетиформным дерматитом Дюринга, простым герпесом.
Лечение синдрома Лайелла
Лечение пациентов с токсическим эпидермальным некролизом проводится в реанимационном отделении и включает целый комплекс неотложных мероприятий. При этом, учитывая токсико-аллергический характер заболевания, применение лекарственных препаратов должно проводиться со строгим учетом показаний и противопоказаний.
Терапия синдрома Лайелла осуществляется инъекционным введением больших доз кортикостероидов (преднизолон). При улучшении пациент переводится на прием препарата в таблетированной форме с постепенным понижением его дозы. Применение методов экстракорпоральной гемокоррекции (плазмаферез, гемосорбция) позволяет производить очищение крови от образующихся при синдроме Лайелла токсических веществ. Постоянная инфузионная терапия (физ. раствор, декстран, солевые растворы) направлена на борьбу с обезвоживанием и нормализацию водно-солевого баланса. Она проводится при строгом контроле объема выделяемой пациентом мочи.
В комплексной терапии синдрома Лайелла применяют медикаменты, поддерживающие работоспособность почек и печени; ингибиторы ферментов, участвующих в разрушении тканей; минеральные вещества (калий, кальций и магний); препараты, снижающие свертываемость; мочегонные средства; антибиотики широкого спектра.
Местное лечение синдрома Лайелла включает применение аэрозолей с кортикостероидами, влажно-высыхающих повязок, антибактериальных примочек. Она проводится в соответствии с принципами обработки ожогов. Для профилактики инфицирования при синдроме Лайелла необходимо несколько раз в сутки проводить смену нательного белья на стерильное, осуществлять обработку не только кожи, но и слизистых. Учитывая выраженную болезненность, местная терапия должна проводиться при соответствующем обезболивании. При необходимости проведение перевязок осуществляется под наркозом.
Прогноз
Прогноз заболевания определяется характером его течения. В связи с этим выделяют 3 варианта течения синдрома Лайелла: молниеносное с летальным исходом, острое с возможным летальным исходом при присоединении инфекционного процесса и благоприятное, обычно разрешающееся спустя 7-10 дней. Ранее начало лечебных мероприятий и их тщательное проведение улучшают прогноз заболевания.
1. Синдром Лайелла как редкое осложнение медикаментозной терапии (клинические случаи)/ Тезяева С.А., Млинник Р.А., Дегтярева С.Ф., Вагапова Т.В, Никольский В.О.// МедиАл. - 2015 - №2 (16).ь
2. К проблеме лечения синдрома Лайелла: вопросы дискутабельного характера/ Владыка А.С. и др.// Украинский журнал дерматологии, венерологии и косметологии. - 2007.
3. Синдром Лайелла (клиника, диагностика, современные методы лечения)/ Чичерина Е.Н. Малых Св. Акшенцева М.В.// Вятский медицинский вестник. - 2008.
Буллезный эпидермолиз
Буллезный эпидермолиз - группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий - «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.

Буллезный эпидермолиз - это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.

Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса - десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии - пограничного буллезного эпидермолиза - являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз - имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз - делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз - имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи - эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе - киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно - развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты - механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств - типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность - как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
ЭПИДЕРМОЛИЗ БУЛЛЕЗНЫЙ
ЭПИДЕРМОЛИЗ БУЛЛЕЗНЫЙ (epidermolysis bullosa; греческий epi- на, поверх + derma кожа + lysis разрушение, растворение; латинский bulla водяной пузырь; синоним: врожденный буллезный эпидермолиз, врожденная пузырчатка, буллезный акантолиз, врожденная буллезная дистрофия, травматический буллезный дерматоз) — редкое наследственное хроническое заболевание кожи, характеризующееся возникновением пузырей в местах, чаще подвергающихся давлению и трению.
Различают две основные формы эпидермолиза буллезного — дистрофическую, описанную Фоксом (W. Т. Fox) в 1879 году, и простую, описанную Гольдшейдером (A. J. Goldscheider) в 1882 году. Простая форма эпидермолиза буллезного отмечается чаще и наследуется по аутосомно-доминантному типу; дистрофическая форма эпидермолиза буллезного может иметь аутосомно-доминантный или аутосомно-рецессивный тип наследования.
При дистрофической форме заболевания пузыри располагаются преимущественно между эпидермисом и дермой (субэпидермально), при простой форме — внутриэпидермально в мальпигиевом слое эпидермиса (шиповатый и базальный слои эпидермиса), реже в роговом. Они возникают в результате дезинтеграции клеток базального слоя эпидермиса, в которых формируются крупные вакуоли, сливающиеся затем во внутриэпидермальные пузыри. Под пузырем сосочки дермы сглажены. При обеих формах в верхней части дермы кровеносные и лимфатические сосуды расширены, отмечается отечность ткани и диффузный, преимущественно лимфоцитарный, инфильтрат разной интенсивности. При дистрофической форме эластические волокна в дерме обычно разрежены или полностью отсутствуют, причем в атрофических участках, остающихся на месте высыпаний, они в дальнейшем не восстанавливаются. В ряде случаев изменения эластических волокон определяются во внешне не измененной коже. При исследовании уртикароподобных рубцов, иногда образующихся на коже при дистрофической форме без предшествующих клинически заметных пузырей, субэпидермально обнаруживаются микропузыри.
Как простая, так и дистрофическая форма заболевания начинаются обычно после рождения и выраженного развития достигают, когда ребенок начинает ползать и ходить. Пузыри разной величины появляются на внешне не измененной коже, главным образом на разгибательных поверхностях конечностей, особенно в области суставов, а также на кистях, стопах, шее, пояснице. Они наполнены прозрачным или геморрагическим содержимым, их появление сопровождается незначительными воспалительными явлениями. Через несколько дней пузыри вскрываются,образуя эрозии, или ссыхаются в корки.
При простой форме эпидермолиза буллезного эрозии на местах пузырей быстро эпителизируются. Обычно к периоду полового созревания процесс затихает. Абортивной разновидностью простой формы является эпидермолиз буллезный Вебера — Коккейна, характеризующийся поражением только стоп (иногда кистей), рецидивирующий в теплое время года и сопровождающийся местным гипергидрозом.
При дистрофической форме, которая протекает более тяжело, после отпадения корок на месте ссохшихся пузырей остаются атрофические рубцы, на фоне которых развиваются эпидермальные кисты, напоминающие милиум (см.); наблюдается дистрофия ногтей (см.), возможны сращение пальцев, резорбция костной ткани отдельных фаланг, деформация кистей и стоп, иногда в сочетании с дистрофией зубов и волос (полидиспластическая разновидность дистрофической формы эпидермолиза буллезного). Симптом эпидермальной отслойки (отслойка эпидермиса в краевой зоне эрозии при потягивании пинцетом за покрышку пузыря) часто положительный. Примерно у половины больных пузыри возникают на слизистой оболочке полости рта, особенно на языке. На этих местах формируются грубые деформирующие рубцы, на которых вновь могут возникать пузыри. Наиболее активно процесс протекает в раннем детском возрасте и в возрасте 12—15 лет.
Разновидностью дистрофического эпидермолиза буллезного являются альбопапулоидный эпидермолиз буллезный, проявляющийся своеобразными белыми уртикароподобными рубцами на месте бывших пузырей, но иногда возникающими и самопроизвольно. Описаны также локализованные формы эпидермолиза буллезного. Редкой разновидностью дистрофического эпидермолиза буллезного является язвенно-вегетирующий эпидермолиз буллезный, при котором на месте пузырей появляются стойкие изъязвления с вегетациями. Выделяют также летальную форму эпидермолиза буллезного, которая встречается редко; она возникает в первые часы после рождения и характеризуется отслоением больших участков эпидермиса в местах, подвергающихся травмированию; дети погибают в первые недели или месяцы жизни.
Диагноз устанавливают на основании клинической картины, подтвержденной в сомнительных случаях данными гистологического исследования.
Дифференциальную диагностику проводят с различными формами буллезных дерматозов (см. таблицу к ст. Пузырчатка).
Лечение включает назначение витаминов А, С, Р, группы В, гамма-глобулина, фитина, препаратов кальция, железа, переливания плазмы, при тяжело протекающих дистрофических формах эпидермолиза буллезного показано применение кортикостероидов. Местное лечение заключается во вскрытии пузырей и нанесении на эрозивные поверхности дезинфицирующих и эпителизирующих мазей.
Прогноз в отношении жизни, как правило, благоприятный. При дистрофической форме в тяжелых случаях может наступить инвалидность в связи с грубой деформацией кистей и стоп, резорбцией костной ткани фаланг, сращением пальцев и др.
Профилактика обострения процесса заключается в предохранении кожи больного от травм, а также от присоединения вторичной инфекции.
Библиогр.: Дифференциальная диагностика кожных болезней, под ред. А. А. Студницина, М., 1983; Лелис И. И. и Лелиене В. А. Наследственный буллезный эпидермолиз и его варианты, Вестн. дерм, и вен., № 6, с. 63, 1977; Машкиллейсон Л. Н. Частная дерматология, с. 268, М., 1965; Самцов В. И. Альбопапулоидный буллезный эпидермолиз, Вестн. дерм, и вен., № 6, с. 64, 1971; Студницин А. А. и др. Клиника и течение буллезного эпидермолиза у детей, там же, № 4, с. 3, 1974; Dermatology in general medicine, ed., by Т. B. Fitzpatrick a. o., p. 334, N. Y., 1979.
Читайте также:
- КТ, МРТ при плоскоклеточном раке подсвязочного отдела гортани
- Требования к питьевой воде (водоснабжению) на предприятии
- Лечение остановки дыхания во сне (синдрома обструктивного апноэ во сне, СОАС)
- Буферная функция барорецепторов. Механизмы поддержания давления барорецепторами
- Перелом нижней челюсти: причины, симптомы и лечение