Синдром расщелины черепа у ребенка
Добавил пользователь Morpheus Обновлено: 21.01.2026
Практически любой врач курса общей анатомии помнит о существовании специфических форм черепа, таких, как скафоцефалический (вытянутый в передне-заднем направлении) и брахицефалический (увеличенный в ширину). Но редко кто вспоминает о том, что необычная форма черепа у ребенка во многих случаях является признаком преждевременного заращения черепных швов.
Конечно, все доктора знакомы с термином краниостеноз - преждевременное заращение швов черепа, приводящее к неспецифическому повреждению головного мозга вследствие недостаточного расширения полости черепа в период наиболее активного роста мозга. Когда возникает вопрос о том, как лечат краниостеноз немногие вспоминают о возможности хирургического иссечения преждевременно заросших швов и лишь единицы знают о существовании метода двухлоскутной краниотомии.
Между тем, по международной статистике, преждевременное закрытие одного из швов черепа (изолированный краниосиностоз) возникает примерной у одного из 1000 детей. Интересно отметить, что такая же частота характерна и у детей, имеющих расщелину губы. При этом ни у кого не вызывает трудностей диагностика расщелин губы, потому что таких пациентов, несомненно, видел каждый врач. Тогда как почти никто из врачей общей практики не может припомнить, видел ли он когда-нибудь ребенка с преждевременным заращением швов черепа.
Хождение по мукам
Краниосиностоз - преждевременное заращение одного или нескольких швов черепа, приводящее к формированию характерной деформации головы. Таким образом, уже в родильном доме ребенок с подозрением на краниосиностоз может быть выделен из общей массы новорожденных и направлен на дообследование. На практике, к сожалению, на этом этапе все деформации черепа, обнаруженные у детей, расцениваются врачами как особенности послеродовой конфигурации головы и им не уделяется должного внимания. В период новорожденности форме черепа также не придается большого значения. Обычный ответ педиатра на обеспокоенность родителей: «…Ничего страшного, хорошо прибавляет в весе, ушла желтуха, а голова такая оттого, что лежит на боку. Вот начнет ходить, и все исчезнет».
Психомоторное развитие детей проходит с отставанием, деформации черепа самопроизвольно не исчезают, некоторые деформации становятся менее заметными, скрываясь под волосами, другие ошибочно расцениваются врачами как иные заболевания, а третьи отступают на второй план при наличии более очевидных нарушений функции органов и систем. Зачастую малышей с краниосиностозами консультируют генетики, и нередко правильно устанавливается группа заболеваний и даже предполагается непосредственный генетический синдром. Несмотря на это, единицы таких пациентов поступают в специализированные клиники для проведения лечения. Подавляющее большинство детей, не получивших лечения, имеют сниженный интеллект и становятся инвалидами. Из-за необычной формы черепа нарушаются пропорции лица, и к периоду полового созревания у таких детей чаще, чем у других, возникают трудности в социальном общении и даже возможны суицидальные попытки.
Родители постепенно перестают обращать внимание на легкие деформации черепа, а при наличии выраженной у ребенка деформации лица детские хирурги разъясняют им, что исправление косметических дефектов проводится только в 16 лет.
Диагностика
Рисунок 1
Основными швами свода черепа являются сагиттальный, коронарный, лямбдовидный и метопический (рис. 1). При преждевременном заращении костного шва происходит компенсаторный рост костей перпендикулярно к его оси (закон Вирхова). В результате появляется характерная деформация. Опишем наиболее часто встречающиеся формы краниосиностозов.
Сагиттальный краниосиностоз
Преждевременное заращение сагиттального шва приводит к увеличению передне-заднего размера черепа с нависающими лобной и затылочной областями и к уменьшению его ширины с формированием узкого овального лица (рис. 2). Такой вид деформации называют скафоцефалией, или ладьевидным черепом. Это наиболее частое заболевание среди общего числа изолированных синостозов (50-60%). Характерная форма черепа видна уже с рождения. При осмотре головы сверху заметно втяжение теменных областей, это дает ощущение циркулярной перетяжки свода черепа на уровне или чуть кзади от ушных раковин. Четко определяется большой родничок, причем его размеры не отличаются от нормы. Характерным считается наличие костного гребня, пальпируемого в проекции сагиттального шва.
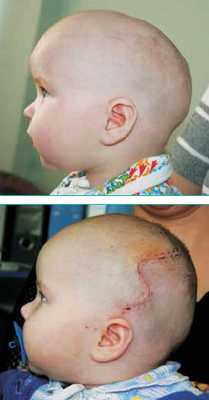
Рисунок 2: а - ребенок до и б - после устранения скафоцефалии.
Метопический краниосиностоз
Самым редким представителем группы изолированных краниосиностозов является метопический краниосиностоз, или тригоноцефалия, составляющая 5-10% от общего их количества. Несмотря на это, данное заболевание, пожалуй, чаще всего распознается как врожденная деформация черепа из-за характерной клинической картины.
При раннем замыкании метопического шва происходит формирование треугольной деформации лба с образованием костного киля, идущего от надпереносья до большого родничка. При взгляде на такой череп сверху видна четкая треугольная деформация с вершиной в области надпереносья. При этом верхние и латеральные края орбит смещаются кзади, что дает ощущение разворота плоскости орбит кнаружи и уменьшения межорбитального расстояния (гипотелоризм). Деформация лба настолько необычна, что дети с тригоноцефалией часто обследуются у генетиков и наблюдаются как носители наследственных синдромов, сопровождаемых снижением интеллекта. Действительно, тригоноцефалия рассматривается как неотъемлемая часть таких синдромов, как Opitz, Oro-facio-digital syndrom, и некоторых других. Верно и то, что многие синдромальные заболевания приводят к задержке интеллектуального развития, но частота их настолько низка, а клиническая картина настолько характерна, что не стоит всех детей, имеющих лишь метопический синостоз, причислять к группе риска по развитию умственной неполноценности.
Односторонний коронарный краниосиностоз
Коронарный шов расположен перпендикулярно срединной оси черепа и состоит из двух равноценных половин. Так что при преждевременном заращении одной из его половин формируется типичная асимметричная деформация, именуемая плагиоцефалией. Вид ребенка с плагиоцефалией характеризуется уплощением верхнеорбитального края орбиты и лобной кости на стороне поражения с компенсаторным нависанием противоположной половины лба (малыш как будто хмурится одной стороной лица). С возрастом более отчетливо начинает проявляться ипсилатеральное уплощение скуловой области и искривление носа в ту же сторону. В школьном возрасте присоединяется деформация прикуса, связанная с увеличением высоты верхней челюсти и как следствие - смещением нижней челюсти на стороне преждевременно закрывшегося шва. В тяжелых случаях имеется даже компенсаторное выбухание затылочной области со стороны синостоза. Нарушения со стороны органа зрения представлены чаще всего односторонним косоглазием. Плагиоцефалия чаще других расценивается как особенности послеродовой конфигурации головы. Но в отличие от последней она не исчезает в первые недели жизни, а, наоборот, с возрастом прогрессирует.
Таким образом, огромную роль в правильной постановке диагноза играет именно форма черепа.
Из инструментальных методов диагностики наилучшим является проведение компьютерной томографии с трехмерным ремоделированием изображения костей свода черепа и лица. Это обследование помогает выявить сопутствующую патологию головного мозга, подтвердить наличие синостоза в случае изолированного повреждения и установить все заинтересованные швы в случае полисиностоза.
Лечение
Самым активным периодом роста головного мозга считается возраст до двух лет. Таким образом, с функциональной точки зрения предотвратить краниостеноз можно ранним хирургическим лечением. Оптимальным возрастом для проведения операции по поводу краниосиностоза можно считать период с 3 до 9 месяцев. Преимуществами лечения в данном возрасте можно считать:
- легкость манипулирования с тонкими и мягкими костями черепа;
- облегчение окончательного ремоделирования формы черепа быстро растущим мозгом;
- более полное и быстрое заживление остаточных костных дефектов.
Если лечение выполняется после пяти лет, сомнительно, что оно приведет к значительному улучшению функции головного мозга. В большей степени операция будет направлена на устранение деформации головы.
Основной особенностью современного хирургического лечения является не только увеличение объема черепа, но и исправление его формы и сочетанной деформации лица в ходе одной операции.
Сведения об авторах:
Андрей Вячеславович Лопатин, зав. отделением челюстно-лицевой хирургии ГУ «Российская детская клиническая больница» Росздрава, профессор, д-р мед. наук
Сергей Александрович Ясонов, врач отделения челюстно-лицевой хирургии ГУ «Российская детская клиническая больница» Росздрава
Шизэнцефалия
Шизэнцефалия — врожденный порок ЦНС в виде расщелины головного мозга, возникающий в результате поздней нейрональной миграции. Основными факторами риска болезни служат генетические дефекты, тератогенные влияния в антенатальном периоде, внутриутробная гипоксия и нейроинфекции. Состояние проявляется полиморфными судорожными приступами, задержкой психомоторного развития, очаговой неврологической симптоматикой. Пренатальная диагностика выполняется на плановом УЗ-скрининге беременности, постнатальная — с помощью церебрального МР-сканирования, нейросонографии, ЭЭГ. Лечение поддерживающее: антиконвульсанты, ноотропы, проведение комплексной реабилитации. При наличии осложнений выполняются нейрохирургические вмешательства.
МКБ-10
Общие сведения
Первые описания патологии были сделаны Вилмартом еще в 1887 году, после чего в 1946 г. ученые Яковлев и Вадсфорф выделили два морфологических подтипа заболевания. Первое описание ультразвуковой картины порока принадлежит У. Клингенсмиту, Д. Койффи-Рагану. Шизэнцефалия встречается с частотой 1,5:100000 живорожденных новорожденных. Намного чаще патология регистрируется среди пациентов с эпилептическими приступами — 1 случай на 1650 больных. Расовых и половых различий среди заболевших не выявлено.
Причины
Этиологические факторы аномалии изучены недостаточно. В современной неврологии существуют разные мнения по поводу порока: одни специалисты связывают его развитие с церебральной ишемией в эмбриональном периоде, другие — с дизрупцией (деструктивным процессом в первоначально правильно сформированном органе), возникшей под влиянием неизвестных причин. Выделяют следующие факторы риска формирования расщелины мозга:
- Генные мутации. У ряда новорожденных присутствует мутантный ген EMX2 (ген гомеобокса), который участвует в росте и дифференцировке нейробластов. Изредка шизэнцефалия выявляется при мутации COL4A1. Поскольку генный дефект есть не у всех пациентов, в этиопатогенезе играют роль и другие факторы.
- Внутриутробная гипоксия. Такое состояние встречается при наличии у матери экстрагенитальных заболеваний (анемия, сердечная недостаточность, болезни легких), патологий беременности (резус-конфликт), различных генетических синдромах у плода.
- Токсические воздействия. Нарушения эмбрионального развития часто провоцируются негативным влиянием табачного дыма (в том числе при пассивном курении), алкоголя, бытовых и промышленных химических токсинов. Некоторые лекарства (антикоагулянты, антибиотики, цитостатики) также могут нарушать формирование ЦНС у плода.
- Внутриутробные инфекции. Возникновение шизэнцефалии возможно при первичном заражении или активации латентной инфекции в первом триместре беременности. Наибольшей тропностью к нервной ткани обладают вирусы простого герпеса, цитомегаловирус, токсоплазма.
Патогенез
Ряд исследователей основным звеном патогенеза называют внутриутробный инсульт в бассейне средней мозговой артерии, что приводит к образованию участка ишемизированной ткани, в котором нарушаются процессы структурной организации клеток. Церебральной гипоксии при формировании шизэнцефалии обычно сопутствует хронический воспалительный процесс, вызванным нейроинфекцией.
Существует два типа болезни с учетом патоморфологических особенностей. Для 1 типа характерна сомкнутая расщелина, края которой разделены узкой бороздкой, покрытой сверху эпендимой и паутинной оболочкой. При 2 типе патологии формируется разомкнутая расщелина с далеко отстоящими друг от друга краями. При этом желудочки сообщаются с субарахноидальным пространством, происходит циркуляция ликвора.
Макроскопически патология у новорожденных представляет собой щель головного мозга с преимущественной локализацией в области латеральной борозды. Микроскопически при таком пороке ЦНС наблюдается дисплазия серого вещества, покрывающего расщелину, нарушение структуры слоев мозговой коры в зоне поражения. Зачастую, кроме шизэнцефалии, у новорожденных, детей другого возраста выявляются другие пороки: врожденные аномалии прозрачной перегородки, мозолистого тела, зрительного перекреста.
Симптомы шизэнцефалии
Клиническая картина заболевания варьирует в зависимости от локализации и размеров расщелины. Средний возраст манифестации симптоматики составляет 4 года, хотя в тяжелых случаях отдельные признаки выявляются уже у новорожденных. Для шизэнцефалии, как и для других кистозных полостей головного мозга, типично запаздывание клинических проявлений, что затрудняет диагностику, если она не была выполнена в ходе пренатального скрининга.
Основным признаком болезни являются разнообразные эпилептические пароксизмы: сложные фокальные приступы (нарушения сознания в сочетании с подергиванием одной из конечностей), простые фокальные припадки, фокальные пароксизмы с вторичной генерализацией. Интенсивность пароксизмальных явлений колеблется от единичных приступов до 10-30 пароксизмов в сутки. У новорожденных и детей первого года жизни отмечаются единичные тонические или миоклонические приступы.
К непароксизмальным симптомам болезни относят нарушения иннервации лицевой мускулатуры, что проявляется асимметрией лица, обеднением мимики. Также развиваются парезы мышц, иннервируемых бульбарной группой нервов, что манифестирует расстройствами глотания, произношения звуков. У новорожденных, а также младенцев возможна персистенция безусловных рефлексов, у детей постарше — гемипаретическая форма ДЦП.
Более редкими признаками расщелины мозга служат нарушения сна, ощущения шума или пульсации в голове, расстройства зрения (яркие вспышки света перед глазами, ухудшение остроты зрения, сужение зрительных полей). Такие жалобы в основном предъявляют дети школьного возраста. Возможны нарушения координации движений, неустойчивость походки, мелкоразмашистый тремор.
Осложнения
Расщелина мозга чревата формированием гидроцефалии, которая проявляется увеличением и деформацией головы ребенка, нарушением оттока ликвора. Без своевременной медицинской помощи существует риск гипертензионно-гидроцефального синдрома. У новорожденного, младенца возникают генерализованные судороги, выбухание родничка, церебральная рвота. При поражении ствола мозга на фоне гидроцефалии возможен летальный исход.
Тяжелый неврологический дефицит наблюдается при сочетании шизэнцефалии и септо-оптической дисплазии, гетеротопии серого вещества. Дети с расщелиной головного мозга страдают от умственной отсталости, задержки речевой функции, различных психических отклонений. Опасность представляют фармакорезистентные судорожные приступы, которые чреваты развитием эпилептического статуса.
Диагностика
На современном этапе развития ультразвуковой диагностики в большинстве случаев шизэнцефалия диагностируется антенатально во время планового скрининга беременности. Намного лучше визуализируется расщелина сомкнутого типа, тогда как открытая шизэнцефалия может остаться незамеченной. Для постнатального подтверждения диагноза детскому неврологу требуются следующие методы исследования:
- МРТ головного мозга. Магнитно-резонансная томография — безопасный метод даже для новорожденных. При 1 типе аномалии обнаруживается выпячивание стенки желудочков с узким каналом. 2 тип характеризуется открытой расщелиной, которая заполнена цереброспинальной жидкостью. При необходимости выполняется МРТ мозговых сосудов.
- Нейросонография. УЗИ головного мозга используется как альтернатива МРТ у новорожденных, младенцев до закрытия большого родничка. Сонография достаточно информативна при шизэнцефалии I типа. Метод также применяется для выявления сопутствующих аномалий строения церебральных структур. Для уточнения диагноза нейросонография дополняется УЗДГ церебральных сосудов.
- ЭЭГ. По результатам электроэнцефалографии у больных разных возрастных групп, включая новорожденных, определяется замедление основной фоновой активности, региональные эпилептиформные паттерны в лобно-центрально-височной зоне. Реже наблюдается вторичная билатеральная синхронизация, мультифокальная эпилептиформная активность.
- Офтальмоскопия. При исследовании глазного дна обнаруживается важный офтальмологический симптом гидроцефалии — отечность дисков зрительных нервов. Метод применим для больных разного возраста, в том числе, новорожденных. Для оценки работы зрительного анализатора рекомендованы визометрия, периметрия, биомикроскопия глаза.
- Генетическое тестирование. Поскольку аномалия сочетается с мутациями EMX2, COL4A1, показано проведение автоматического секвенирования экзона. Однако информативность такого метода остается невысокой ввиду полиэтиологичности заболевания.
Лечение шизэнцефалии
Консервативная терапия
Медицинская помощь детям с шизэнцефалией ограничивается поддерживающим симптоматическим лечением. Больным показано динамическое наблюдение у детского невролога, регулярный КТ- или МРТ-контроль состояния расщелины, определение наличия или отсутствия вторичных структурных изменений мозговых тканей. Основные группы препаратов, которые включаются специалистом в индивидуальную схему лечения шизэнцефалии:
- Антиконвульсанты. Назначаются различные комбинации противосудорожных препаратов для купирования эпилептических приступов, предупреждения рецидивов.
- Нейрометаболиты. Препараты, улучшающие трофику головного мозга, используются для стимуляции психоречевого и моторного развития, уменьшения неврологического дефицита.
- Дегидратанты. Для нормализации ликвородинамики могут рекомендованы осмотические диуретики, салуретики, онкодегидратанты. Лечение проводится под контролем диуреза и объема вводимых инфузионных растворов.
Оперативное лечение
Хирургическое лечение применяется при осложненном течении заболевания. Обычно операции проводятся новорожденным, младенцам для ликвидации нарастающей гидроцефалии. Чаще всего выполняются эндоскопическая вентрикулостомия III желудочка и ликворошунтирующие вмешательства. При учащении эпилептических пароксизмов, появлении и нарастании у новорожденного либо пациента другого возраста грубой очаговой неврологической симптоматики производится аспирация содержимого крупных кист.
Реабилитация
Важную роль играет комплексная реабилитация пациентов, включая новорождённых и детей младенческого возраста. Для повышения мышечной силы, объема произвольных движений используется массаж, рефлексотерапия, механотерапия и ЛФК. Для развития речи ребенку требуется помощь логопеда-дефектолога. С целью повышения обучаемости и социализации больных прибегают к услугам нейропсихолога, коррекционного педагога.
Прогноз и профилактика
Благоприятное течение характерно для новорожденных с сомкнутой односторонней шизэнцефалией, которая не сопровождается грубым неврологическим дефицитом. Внушает опасения разомкнутая билатеральная форма порока, вызывающая раннюю инвалидизацию и смерть больных. Профилактические меры включают рациональное ведение беременности, исключение тератогенных влияний на организм будущей матери, усовершенствование методики пренатальных УЗ-скринингов.
1. Шизэнцефалия (обзор литературы и клинические наблюдения/ О.А. Милованова, И.М. Мосин, Л.В. Калинина// Неврологический журнал. — 2011. — №3.
2. Пренатальная диагностика шизэнцефалии/ А.Е. Волков, Е.Н. Андреева// SonoAce Ultrasound. — 2010. — №21.
3. Лучевые методы исследования, МРТ головного мозга у больных с шизэнцефалией/ М.И. Пыков, К.В. Ватолин, О.А. Милованова, Н.В. Чернышева// Эффективная фармакотерапия. Педиатрия. — 2010. — №5.
4. Шизэнцефалия, тип II: случай пренатальной диагностики/ Е.Н. Андреева// Пренатальная диагностика. — 2008. — №4.
Краниосиностоз
Краниосиностоз - это заболевание, основным симптомом которого является деформация мозгового отдела черепа, возникающая вследствие преждевременного зарастания костных швов. Клиника включает в себя деформации черепа, симптомы внутричерепной гипертензии, патологию зрительного нерва, отставание в психическом развитии. Редко заболевание сопровождается аномалиями костей лицевого черепа. Диагностика заключается в оценке степени зарастания черепных швов и определении костных дефектов путем физикального обследования, рентгенографии, КТ и МРТ. Основное лечение - ранняя хирургическая коррекция формы костей черепа.
Краниосиностоз - это патологическое состояние в педиатрии, возникающее на фоне раннего зарастания черепных швов, характеризующееся деформацией черепной коробки и нарушением развития тканей головного мозга. В среднем распространенность разных форм заболевания в станах СНГ составляет 0,03-3,5% от всех новорожденных. Мужской пол более склонен к развитию данной патологии. Наиболее распространенный вариант - моносиностоз. Чаще всего наблюдается преждевременное зарастание сагиттального шва (скафоцефалия) - 50-65% от всех краниосиностозов. Самой редкой и прогностически неблагоприятной является синдромальная форма, при которой имеется высокий риск летального исхода на первом году жизни ребенка. При своевременной диагностике и адекватном лечении в первые 6-9 месяцев жизни дальнейшее развитие пациента проходит без отклонений.
Причины краниосиностоза
Точная этиология краниосиностоза не установлена. Согласно выдвинутым теориям, данное заболевание может развиваться в результате внутриутробного нарушения гормонального фона ребенка, перинатальных травм и сдавливания костей черепа в полости матки. Также данная патология возникает при наследственных патологиях - синдроме Апера, синдроме Крузона и синдроме Пфайффера. Доподлинно известна одна из ведущих причин развития краниосиностоза - аномалия гена, отвечающего за образование рецепторов фактора роста фибробластов (FGFR типы I, II и III).
Патогенетически краниосиностоз обусловлен преждевременным синостозированием одного или сразу нескольких черепных швов: коронарного, сагиттального, лямбдовидного или метопического. На фоне этого, согласно закону Вирхова, возникает компенсаторный рост костной ткани в перпендикулярном направлении, из-за чего формируется деформация черепа. Полисиностоз (а зачастую - и моносиностоз) часто сопровождается внутричерепной гипертензией, которая может проявляться неврологическими нарушениями вследствие сдавливания коры головного мозга, венозным застоем глазного дна, отеком диска зрительного нерва, а при длительном течении - полной атрофией зрительного нерва и потерей зрения.
Классификация краниосиностоза
Краниосиностоз, согласно этиологическим факторам, разделяют на две группы:
- Синдромальный. В данном случае патология сочетается с другими врожденными пороками. Сюда относятся сцепленные с Х-хромосомой, моногенные, хромосомные и другие краниосиностозы. Например - комбинация синостоза с дисплазией костей лицевого черепа, синдром Смита-Лемли-Опица или рото-пальце-лицевой синдром.
- Несиндромальный. Это изолированная форма, которая возникает самостоятельно и не имеет сопутствующих заболеваний.
В зависимости от количества заросших черепных швов выделяют:
- Моносиностоз. Характеризуется поражением только 1 шва. В случае с коронарным и лямбдовидным швом зарастание может быть одно- или двухсторонним. Наиболее распространенная форма.
- Полисиностоз. В патологический процесс втягиваются 2-3 шва.
- Пансиностоз. При этой форме наблюдается сращивание всех костных швов черепа ребенка. Встречается крайне редко.
Симптомы краниосиностоза
Клинически краниосиностоз проявляется с момента рождения ребенка. Для всех форм характерны плагиоцефалия и раннее закрытие большого родничка (в норме это происходит в 12-18 месяцев). Только при полисиностозе или сопутствующей гидроцефалии он может оставаться открытым до 3-х летнего возраста. Также при краниосиностозах зачастую наблюдается повышение внутричерепного давления, которое может проявляться неврологическими нарушениями: беспокойством, интенсивным плачем, тошнотой и рвотой, нарушением сна, снижением аппетита, позитивным симптомом Грефе, судорогами.
Каждая из форм заболевания имеет характерные клинические особенности. Краниосиностоз стреловидного шва (скафоцефалия или ладьевидный череп) характеризуется увеличением переднезаднего размера головы ребенка при недостаточности ее ширины. Визуально определяется вытягивание черепа, «вдавливание» височных областей, «нависание» лба и затылочной части, сужение лица и приобретение им овальной формы. Пальпаторно над местом прохождения стреловидного шва выявляется костный гребень. В раннем возрасте возможна задержка психического развития.
Зарастание лямбдовидного шва чаще всего носит односторонний характер и проявляется уплощением затылочной области. Является трудно диагностируемой формой, поскольку плагиоцефалия практически незаметна под волосами, а неврологические нарушения минимальны. При взрослении пациента динамика заболевания практически отсутствует.
Коронарный или венечный краниосиностоз может быть как одно-, так и двухсторонним. Зарастание только одной половины шва сопровождается типичной деформацией черепа ребенка - уплощением лобной кости и верхней части глазницы с пораженной стороны. При этом противоположная половина компенсаторно «нависает». Со временем развиваются искривление носа в противоположную сторону, уплощение скулы, нарушение прикуса и косоглазие. Двухсторонний коронарный краниосиностоз проявляется широким, плоским и высоким лбом с уплощенными глазничными краями лобной кости, редко - башенной деформацией черепа (акроцефалией). Неврологические нарушения неспецифичны и аналогичны другим формам.
Нетопический краниосиностоз или тригоноцефалия характеризуется развитием треугольного лба с костным килем, проходящим от глабеллы до большого родничка. Также наблюдается гипотелоризм - смещение глазниц кзади с уменьшением межглазничного промежутка. Со временем происходит некоторое сглаживание костного гребня и нормализация формы лба. В половине случаев возникают нарушения зрения и отставание в психическом развитии.
Синдромальный краниосиностоз является самой редкой и тяжелой формой. Помимо плагиоцефалии отмечается дисплазия костей лицевой части черепа, из-за чего возникают дыхательная недостаточность, нарушение приема пищи и патология зрения. Характеризуется синостозом венечного шва и, как результатом - брахицефалической формой головы ребенка. Также возникают гипоплазии костей верхней челюсти, выпячивание глазных яблок из орбит, гипертелоризм. Часто наблюдается значительное расширение родничка и расхождение стреловидного шва. Без лечения у детей развивается выраженное отставание в психическом развитии, зачастую они погибают на протяжении первых 12 месяцев жизни от ОРВИ, осложнившихся пневмонией.
Диагностика краниосиностоза
Диагностика краниосиностоза базируется на физикальном осмотре и инструментальных методах исследования. Анамнез нередко малоинформативен, но его данные позволяют педиатру проследить динамику клинической симптоматики, если таковая имеет место. Важным моментом становится визуальный осмотр ребенка, который дает возможность обнаружить характерные деформации черепа, аномалии костей и т. д. Лабораторные анализы специфических изменений не выявляют и могут использоваться с целью определения генетической патологии или диагностики осложнений.
Обязательными являются инструментальные методы, позволяющие визуализировать костные деформации и оценить степень поражения тканей головного мозга. Сюда относятся нейросонография, рентгенография, компьютерная и магнитно-резонансная томография. Нейросонография используется с целью оценить состояние тканей головного мозга и размеры желудочков, выявить внутричерепную гипертензию. На рентгенограмме удается определить нарушения структуры костей, окостенение черепных швов, а при повышенном внутричерепном давлении - усиление пальцевых вдавлений. КТ и МРТ применяются для получения более информативных результатов. При подозрении на поражение зрительной системы проводится офтальмоскопия, позволяющая обнаружить поражение диска зрительного нерва. Рекомендованы консультации нейрохирурга и офтальмолога.
Дифференциальная диагностика краниосиностоза осуществляется с позиционной плагиоцефалией, родовой травмой новорожденных (кефалогематомой, подапоневротическим кровоизлиянием, переломом костей черепа), кистами головного мозга, рахитом и микроцефалией.
Лечение краниосиностоза
Основное лечение краниосиностоза - хирургическая коррекция костной деформации черепа. Оптимальное время для проведения оперативного вмешательства - первые 6-9 месяцев жизни ребенка. Данные сроки обусловлены тем, что в этом периоде наблюдается наиболее интенсивное развитие тканей головного мозга, которому может препятствовать деформация черепной коробки. Кроме того, кости черепа в этом возрасте быстро восстанавливают свою структуру без развития осложнений. Объем и техника операции зависят от формы краниосиностоза и сопутствующих патологий. В 2-3-х летнем возрасте коррекция проводится исключительно с целью ликвидировать косметический дефект. Помимо хирургического лечения осуществляется изменение рациона ребенка в соответствии с возрастными требованиями. При развитии интеркуррентных заболеваний показана медикаментозная терапия.
Прогноз и профилактика краниосиностоза
Прогноз для детей с краниосиностозом напрямую зависит от формы заболевания, своевременности диагностики и эффективности оперативного вмешательства. При качественном проведении лечебных мероприятий исход заболевания, как правило, благоприятный. Прогностически неблагоприятной принято считать синдромальную форму краниосиностоза.
Специфической профилактики для данной патологии не существует. Неспецифические меры подразумевают медико-генетическую консультацию семьи и планирование беременности, охрану здоровья женщины при вынашивании ребенка, рациональное питание, отказ от вредных привычек и исключение всех потенциальных этиологических факторов развития краниосиностоза.
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
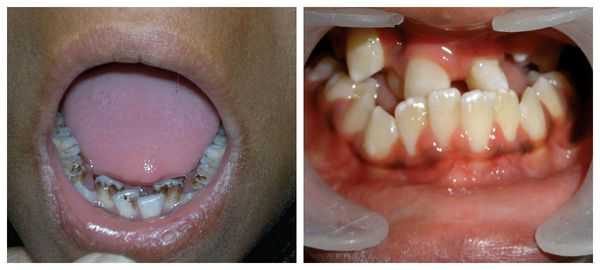
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
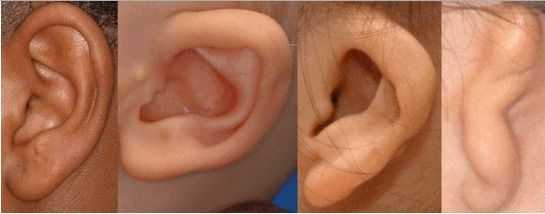
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
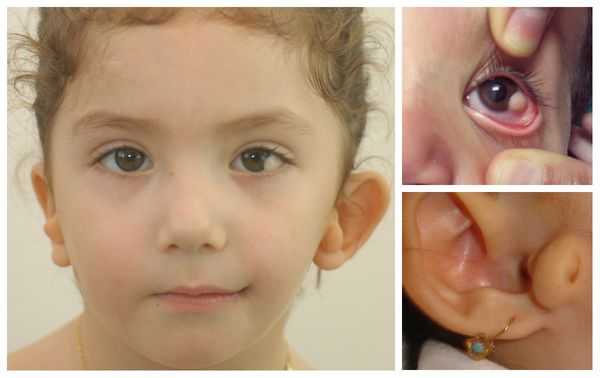
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
Патогенез синдрома Гольденхара
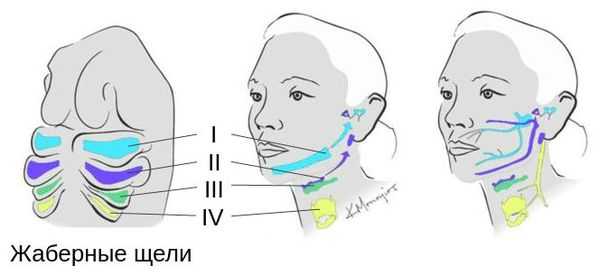
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
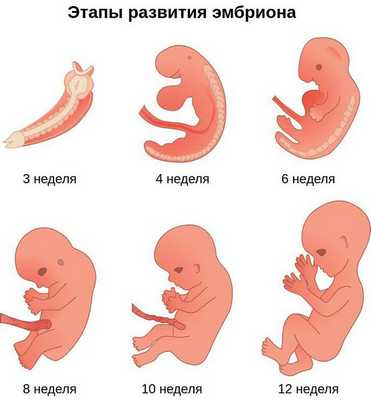
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
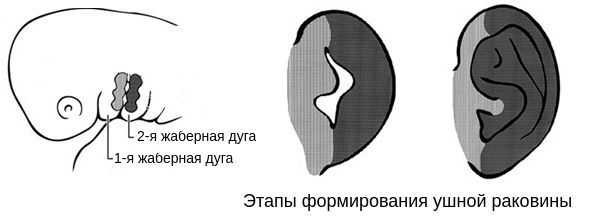
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
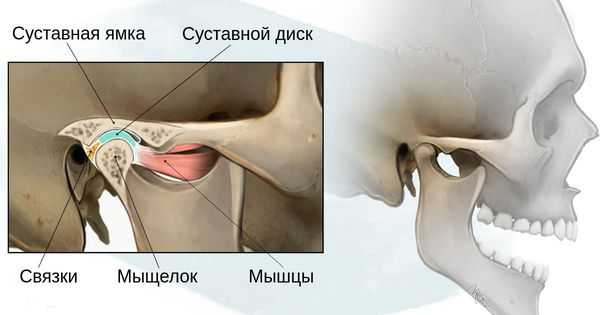
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
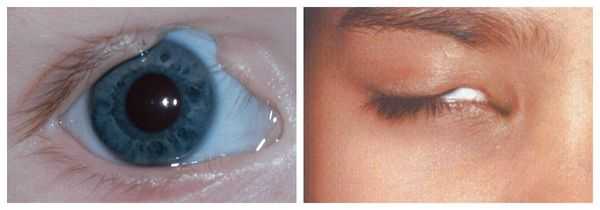
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
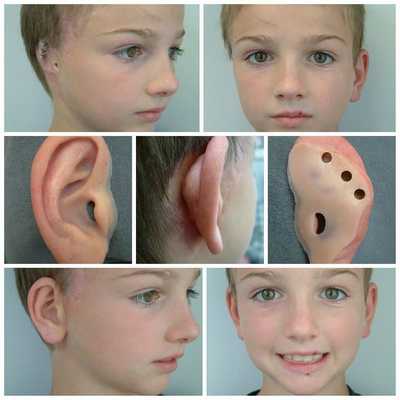
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Врожденная расщелина неба
Врожденная расщелина неба — не такое уж и редкое явление, она встречается примерно у 1 из 500 появившихся на свет детей. Причем, если расщепление губы обычно считается изолированным пороком, расщелина неба может как возникать сама по себе, так и быть частью какого-либо комплексного отклонения, например, последовательность Пьера Робена, синдром Кабуки, синдром Дауна и другие.
Существует очень много форм данного порока: расщелина мягкого неба, расщелина твердого неба, сквозная расщелина, затрагивающая все небо, а также скрытая расщелина, обнаружить которую удается далеко не сразу. В некоторых случаях родители, обеспокоенные плохим развитием ребёнка, медленным набором веса у малыша, грешат на недостаток молока у матери или на качество молочной смеси, пытаются найти тот самый волшебный продукт, который наконец-то поможет ребёнку насытиться, и лишь только рентгеновский снимок позволяет выявить скрытую расщелину, мешающему нормальному сосанию груди или соски.
Причины возникновения расщелины неба
Какой-то одной явной причины для возникновения врожденной расщелины неба нет, обычно этот порок обуславливается стечением сразу нескольких неблагоприятных факторов. Как правило, злоупотребление алкоголем или никотином, на которые принято грешить прежде всего, оказывается совершенно ни при чем. А вот прием определенных медикаментов в ранний период беременности, передозировка витаминов А и Е, воспалительные заболевания на сроке от 5 до 12 недель, стресс, а также наследственный фактор вполне могут воспрепятствовать срастанию костей и мягких тканей лица у эмбриона, в той или иной степени. Немаловажную роль играет и недостаток фолиевой кислоты, врачи подозревают, что нехватка этого витамина вполне может стать причиной возникновения расщелины мягкого и твердого неба.
Где оперировать расщелину неба
Лечение расщелины мягкого неба, равно как и твердого, представляет собой оперативное вмешательство, проводимое в определенные сроки. Еще совсем недавно считалось, что операции на небе стоит делать не раньше 5-6 лет, чтобы рубцы не мешали нормальному росту челюсти, сегодня мнение медиков во всем мире изменилось. Ранние операции необходимы для нормального речевого развития малыша, именно поэтому расщелина мягкого неба может быть прооперирована уже в возрасте от 3 месяцев, а расщелина твердого неба в период от 6 до 24 месяцев.
Лечение врожденной расщелины неба — дело непростое, поэтому лучше доверить его специалисту, проводящему не менее 100 подобных операций в год. Найти такого врача можно в специализированных центрах, информация о которых выложена в интернете, в частности на форуме родительской взаимопомощи Улыбки наших детей.
И не нужно впечатляться народным вариантом названия порока. Тому, кто до сих пор употребляет в своем лексиконе термин «волчья пасть», следует посоветовать забыть об этом термине. Официальное название порока вполне отражает суть явления и не оскорбляет ни родителей, ни самого ребёнка. Врожденная расщелина неба — это всего лишь несращение тканей черепа в период формирования лица ребёнка. Проблема, которая вполне успешно решается, и уж тем более не является причиной для отказа от ребёнка и оставления его в родильном доме.
Читайте также: