Синдром Силвермана (Silverman) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 28.01.2026
Синдром дыхательных расстройств (СДР) — патологическое состояние, возникающее у новорожденных в первые часы после рождения вследствие резкого снижения синтеза сурфактанта и незрелости структуры легочной ткани. Сочетание таких факторов, как недоношенность, внутриутробная инфекция, перинатальная гипоксия и асфиксия, определяют не только причину, но и степень тяжести клинических проявлений СДР. Дефицит сурфактанта — поверхностно-активного вещества, синтезируемого альвеолоцитами II типа, приводит к спадению альвеол на выдохе, в результате чего резко снижается площадь газообмена в легких и как следствие развиваются гипоксемия и гиперкапния. Клинические признаки СДР появляются в первые 4—6 ч после рождения и характеризуются одышкой — более 60 дыхательных движений в 1 мин (причем, чем тяжелее форма заболевания, тем раньше выявляется этот симптом). Учащенное дыхание сопровождается экспираторными шумами. При осмотре - западение грудной клетки на вдохе, отмечается втягивание мечевидного отростка грудины, межреберий, надключичных ямок. Выявляется напряжение крыльев носа, приступы апноэ, появляются цианоз, акроцианоз, общая бледность кожи. Аускультативно ослабленное дыхание. шкала Сильвермана используется для оценки тяжести ДР у новорожденных: (до 4 баллов — начинающийся СДР; 5 баллов — СДР средней тяжести; 6—9 баллов — тяжелый СДР; 10 баллов — крайне тяжелый СДР). С момента появления первых симптомов СДР ребенку начинают проводить оксигенотерапию, цель которой — обеспечение адекватного снабжения тканей кислородом при минимальном риске его токсического действия. Сроки начала энтерального питания определяются в зависимости от состояния ребенка.
Задача: По скорой помощи обратилась 20-летняя женщина, выписанная 2 дня назад из родильного дома. Жалобы на повышение температуры, ознобы, слабость, частое мочеиспускание. Беременность без осложнений. Роды срочные 8 дней назад, осложнились слабостью родовой деятельности и послеродовым кровотечением. Объективно: температура 38,2 *С, пульс - 112 уд. в мин., АД 110/70 мм. рт. ст. Кожные покровы бледные. Живот мягкий, несколько болезненный при пальпации в нижних отделах.Предполагаемый диагноз? Дальнейшая тактика?
Билет 35.
Об этом полезно знать:
Виды упражнений в лёгкой атлетике Характеристика отдельных видов спорта Легкая атлетика - вид спорта, объединяющий естественные для человека физические упражнения.
Расчет коэффициента эластичности спроса по цене 1. Рассчитаем коэффициент эластичности спроса по цене. Р1 = 30 руб.
Порядок приёма и сдачи смены. В каких случаях оператору запрещается сдавать или принимать смену Оператор, принимающий смену, должен явиться на работу за 15 - 20 минут до начала смены и.
Патопсихологические особенности преступников олигофренов Под олигофренией понимают группу заболеваний различной этиологии, общим и типичным для которых является психическое недоразвитие.
Структура системы здравоохранения в России Нормативно-правовые аспекты организации системы здравоохранения Структура системы здравоохранения в России Здравоохранение &ndash.
Для создания семьи достаточно полюбить. А для сохранения — нужно научиться терпеть и прощать. © Мать Тереза ==> читать все изречения.
Синдром Куррарино-Сильвермана. Причины, симптомы, диагностика и лечение
Любая структура и система человеческого организма под действием тех или иных неблагоприятных факторов может развиваться неправильно, аномально. К этому предрасположена даже такая фундаментальная, панцирная часть опорно-двигательного аппарата, как грудная клетка. В качестве самостоятельных синдромов в вертебрологии выделено и описано множество вариантов аномального развития грудной клетки. Одни из них встречаются достаточно часто, другие исключительно редки. В среднем, аномальное строение грудной клетки отмечается примерно у 2% людей.
Наиболее распространенные варианты такой аномалии - вогнутая (воронкообразная) и килевидная (выдающаяся кпереди) грудь. Синдром Куррарино-Сильвермана на сегодняшний день является наиболее редкой, по частоте встречаемости, формой врожденной деформации груди. Данный синдром называют также «бычим рогом» и «верхним килем»: грудная клетка выпячена вперед более или менее острым выступом. Иногда выпячивание кпереди сочетается с воронкообразным вдавлением груди вокруг «киля». Названа данная аномалия в честь ученых, которые первыми дали ее подробное клиническое описание и к 60-м годам ХХ века ввели в медицинскую нозологическую лексику.
2. Причины
Основной причиной развития синдрома Куррарино-Сильвермана считают преждевременное окостенение грудины с гипертрофией 2-4 хрящей и/или нескольких ребер. Причины такого деформирующего разрастания, в свою очередь, до сих пор не прояснены. Установлено, что определенную роль играет наследственность, однако этот фактор удается выявить лишь в 25% случаев. Известно также, что у мальчиков синдром Куррарино-Сильвермана встречается вчетверо чаще, чем у девочек. Исследования продолжаются, однако набор материала и статистический анализ проблематичны из-за редкости заболевания.
3. Симптоматика, диагностика
Выпячивание грудной клетки по типу верхнего киля может быть симметричным и асимметричным, односторонним и двусторонним. Аномалия развития грудной клетки проявляется в детстве и окончательно формируется к пубертатному возрасту. Однако если у маленьких детей такая деформация, как правило, не создает какой либо угрозы внутренним органам, то у более старших пациентов нередко наблюдается сдавление сердца и органов дыхания, одышка, утомляемость из-за постоянного дефицита оксигенации, учащенное дыхание, астма и пр. Кроме того, синдром Куррарино-Сильвермана нередко сопровождается врожденными пороками сердца (например, пролапс митрального клапана). Наконец, нельзя не учитывать влияние отчетливого и достаточно выраженного, в некоторых случаях, эстетического дефекта на развивающуюся психику, что может стать источником серьезных психологических нарушений.
4. Лечение
В разные периоды синдром Куррарино-Сильвермана лечили различными способами. Довольно долго в вертебрологии было распространено мнение, что данную деформацию следует корригировать механически - практиковались специальные сдавливающие корсеты, которые пациент должен был носить постоянно в течение двух лет. Однако к настоящему времени представления о целесообразности такого подхода по ряду причин пересмотрены: постоянное ношение жесткого корсета может быть весьма болезненным и, вместе с тем, неэффективным (несмотря на столь продолжительное и сложное для пациента лечение), а то и вредным для прочих органов. На сегодняшний день методом выбора является хирургическая коррекция. Практикуются различные методики торакопластики - по Равичу, по Кондрашину и пр. Некоторые варианты вмешательства, - напр., металлостернохондропластика Тимощенко, - предполагают имплантацию в грудную клетку специальной корригирующей металлической пластины на срок до полугода, однако такая методика также является дискутабельной.
Оптимальным возрастом операции по устранению верхнего киля считают 14-15 лет. В тех случаях, когда синдром Куррарино-Сильвермана сочетается с врожденными пороками сердца или иными сопутствующими аномалиями, чаще всего целесообразна и показана комбинированная операция: устраняется не только вертебрологическая, но и кардиологическая патология.
Синдром Силвермана (Silverman) - синонимы, авторы, клиника
Институт хирургии им. А.В. Вишневского, Москва
Центральный институт травматологии и ортопедии им. Н.Н. Приорова, Москва
Хирургическая коррекция деформации грудной клетки при синдроме Currarino-Silverman
Журнал: Вестник травматологии и ортопедии им Н.Н. Приорова. 2018;(4): 95‑98
Синдром Currarino-Silverman (комбинированная деформация грудной клетки — ДГК) относится к редким формам ДГК. Часто этот вид деформации сочетается с пороками развития сердца (коарктация аорты, пролапс, стеноз митрального клапана). В статье выделены два типа комбинированной ДГК, что позволяет определять тактику хирургического лечения. Наиболее перспективен с точки зрения возможности оперативной коррекции 2-й тип деформации с протрузией манубриостернального сочленения, но без западения грудины. В работе представлено описание успешно выполненных операций у 3 пациентов с комбинированной ДГК, у одного из которых патология сочеталась с синдромом Поланда.
Синдром Currarino-Silverman, также известный в литературе как комбинированная деформация грудной клетки (ДГК) или особый вид килевидной ДГК (pigeon breast, рectus сarinatum type 2 deformity or upper рectus сarinatum), относится к редким формам ДГК и характеризуется ранним синостозом частей грудины, остановкой роста последней и, как следствие, вторичной ДГК. Это проявляется протрузией манубриостернального сочленения и западением средней и нижней третей грудины. Чаще встречаются симметричные, реже — асимметричные формы комбинированной ДГК. Могут присутствовать и другие сопутствующие нарушения развития опорно-двигательного аппарата, такие как сколиоз, и в редких случаях синдром Поланда 3.
Первым описал комбинированный вид ДГК и предложил методику ее хирургической коррекции М. Ravitch в 1952 г. [4, 5]. Он рассматривал эту деформацию в рамках килевидной ДГК, выделив ее в отдельный вид и назвав «грудью голубя» (pigeon breast). Операция заключалась в субнадхрящничной резекции 2-7 реберных хрящей с обеих сторон, отсечении мечевидного отростка и передней клиновидной стернотомии на уровне наибольшего выстояния грудины. В последующем грудину сшивали на уровне верхней стернотомии, тем самым устраняя ее аркообразную деформацию, опускали до нормального уровня и к передней поверхности нижней трети тела грудины фиксировали мечевидный отросток на мышечной ножке. Дополнительно проводили сборивание перихондра резецированных ребер. Никакие фиксаторы грудинореберного комплекса (ГРК) при этом не использовались.
В 1958 г. G. Currarino и F. Silverman [6] впервые обратили внимание на преждевременное закрытие зон роста грудины. Также они указали на большой процент сочетанных с комбинированным видом ДГК пороков развития сердца (коарктация аорты, пролапс, стеноз митрального клапана) и преобладание в этой категории пациентов женского пола.
С.С. Рудаков и соавт. [1, 7] рассматривают комбинированную ДГК как проявление мальформации или истинного порока развития. На это указывают проявление заболевания сразу после рождения и отсутствие быстрого прогрессирования, т. е. деформация прогрессирует по мере роста ребенка. Основным показанием к операции, по мнению авторов, выступает косметический дефект.
Учитывая травматичность оперативного вмешательства, высокую частоту сочетанных пороков развития сердца, почек и других внутренних органов, операции по исправлению ДГК у больных предпринимаются редко. Показанием к операции в большинстве случаев является косметический дефект.
В настоящем исследовании выделены два типа комбинированной ДГК: 1-й тип — с протрузией манубриостернального сочленения и западением средней и нижней третей грудины и 2-й тип — с протрузией манубриостернального сочленения, без западения грудины. Исходя из типа деформации, выстраивалась хирургическая тактика лечения. При 1-м типе комбинированной ДГК выполняли операцию по методике Ravitch. Более перспективным для хирургической коррекции считается 2-й тип, поскольку в этом случае возможно выполнение менее травматичного хирургического вмешательства. Операция ограничивается только поднадкостничным удалением деформированного фрагмента грудины с замещением его трансплантатом или сшиванием фрагментов грудины.
Представляем собственный опыт хирургического лечения комбинированной ДГК у 3 пациентов.
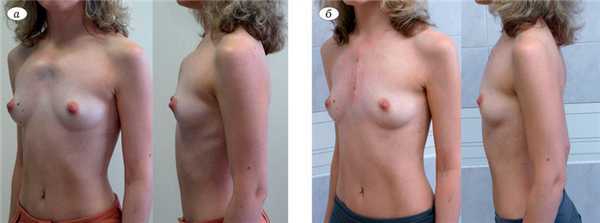
Пациентка Б., 33 лет, в июле 2014 г. поступила в 10-е отделение ЦИТО им. Н.Н. Приорова с жалобами на наличие грубой ДГК и обусловленный этим косметический дефект (рис. 1, а). Рис. 1. Внешний вид пациентки Б., 33 лет, с комбинированной ДГК симметричной формы (2-й тип) до (а) и через 8 дней после (б) операции. ДГК появилась с рождения и прогрессировала пропорционально росту организма. Наследственность не отягощена.
При осмотре, начиная с уровня 2-го ребра, наблюдалось аркообразное выстояние грудины и хрящевых отделов 2-4-го ребер. Средняя и нижняя трети грудины расположены правильно, без тенденции к западению. Реберные дуги не выстоят.
Пациентке провели комплексное предоперационное обследование. Был выявлен пролапс митрального клапана 1-й степени, без регургитации.
Операцию выполнили 08.07.14. По средней линии грудины был осуществлен доступ длиной 10 см. Мобилизовали грудные мышцы в пределах зоны деформации. Провели субнадхрящничную резекцию 2-4-го реберных хрящей с обеих сторон с костной частью, поднадкостничную резекцию деформированной части грудины на протяжении 6 см. В ложе удаленного фрагмента грудины были уложены фрагменты костной ткани. Была восстановлена целостность передней пластинки грудины отдельными швами. Осуществили сборивание перихондра резецированных реберных хрящей по Ravitch. Была достигнута правильная конфигурация грудины. Грудную клетку зафиксировали в корсете. Послеоперационный период протекал без осложнений, на 9-е сутки пациентка была выписана на амбулаторное лечение (см. рис. 1, б).
Пациент М., 17 лет, в июне 2017 г. поступил с жалобами на наличие грубой ДГК и обусловленный этим косметический дефект (рис. 2, а). Рис. 2. Пациент М., 17 лет, с комбинированной ДГК симметричной формы (2-й тип). а — внешний вид до операции; б — рентгенограмма грудной клетки в боковой проекции; в — внешний вид через 3 мес после операции. ДГК с рождения.
При осмотре, начиная с уровня 3-го ребра, наблюдалось аркообразное выстояние грудины и хрящевых отделов 3-5-го реберных хрящей. Нижние отделы грудины без тенденции к западению (рис. 2, б).
В ходе обследования был диагностирован пролапс митрального клапана 1-й степени с умеренной митральной регургитацией.
Операция была проведена 30.06.17. По средней линии грудины выполнили доступ длиной 10 см. Были мобилизованы грудные мышцы в пределах зоны деформации. С техническими сложностями осуществили субнадхрящничную резекцию 3—5-го реберных хрящей с обеих сторон с костной частью, поднадкостничную резекцию деформированной части грудины на протяжении 6 см. Фрагменты грудины сшивали проволокой. Была восстановлена целостность передней пластинки грудины, а также достигнута правильная конфигурация грудины.
Послеоперационный период прошел без осложнений, на 10-е сутки пациент был выписан на амбулаторное лечение. При осмотре через 3 мес: жалоб нет. Констатированы правильная конфигурация грудной клетки и консолидация грудины в зоне остеотомии (рис. 2, в).
Пациент К., 16 лет, в марте 2018 г. поступил в 10-е отделение Центрального института травматологии и ортопедии им. Н.Н. Приорова с жалобами на наличие грубой ДГК и обусловленный этим косметический дефект (рис. 3, а). Рис. 3. Пациент К., 16 лет, с комбинированной ДГК асимметричной правосторонней формы и синдромом Поланда слева. а — внешний вид до операции; б — данные КТ грудной клетки: отмечаются аркообразная, винтообразная деформация грудины, патологически широкая, оссифицированная грудина, аплазия грудных мышц слева; в — внешний вид через 4 мес после операции. Деформация с рождения.
При осмотре: левая половина грудной клетки недоразвита. Большая и малая грудные мышцы слева аплазированы. Начиная с уровня 2-го ребра, наблюдались аркообразное выстояние грудины с винтообразной деформацией и выстояние хрящевых отделов 2-5-го реберных хрящей, больше справа. Нижние отделы грудины без тенденции к западению (рис. 3, б). Реберные дуги не выстоят. Верхние конечности развиты симметрично, функция их не нарушена.
При обследовании сопутствующей патологии выявлено не было. Был поставлен диагноз: комбинированная ДГК, асимметричная правосторонняя форма. Синдром Поланда слева (аплазия большой и малой грудных мышц). Пациента стали готовить к операции.
Операция была выполнена 22.03.18. По средней линии грудины осуществили доступ длиной 10 см. Были мобилизованы грудные мышцы в пределах зоны деформации. Проведена субнадхрящничная резекция 2-7-го реберных хрящей справа и 3-5-го — слева. Ребра со 2-го по 5-е справа были резецированы с костной частью. Осуществили поднадкостничную плоскостную резекцию деформированной части грудины, сборивание перихондра резецированных реберных хрящей. Была достигнута правильная конфигурация костного каркаса грудной клетки.
Согласно линиям разметки, сформирован подкожный карман-ложе, в который установлен пекторальный имплантат (POLYTECH) объемом 231 мл. Грудная клетка приобрела правильную конфигурацию.
Несколько названий одной и той же патологии — синдром Currarino-Silverman, комбинированная ДГК, «грудь голубя» (pigeon breast), килевидная ДГК 2-го типа или верхняя килевидная ДГК — вносят путаницу в литературе, затрудняют осмысление характера патологического процесса и не всегда позволяют принимать правильное решение при лечении пациентов этой категории. На взгляд авторов настоящей статьи, наиболее правильным следует считать термин «комбинированная ДГК». Патологию необходимо рассматривать как истинный порок развития, т. е. ДГК не прогрессирует, а в послеоперационном периоде практически не бывает рецидивов. В синдромальном спектре больных с комбинированной ДГК присутствуют такие синдромы, как Шерешевского-Тернера, Нунан и др. Высокий процент сочетания комбинированной ДГК и пороков развития сердца, почек и ряда других внутренних органов требует комплексного обследования пациентов. Хирургическая коррекция деформации предпринимается по косметическим показаниям при отсутствии у больного пороков развития внутренних органов, требующих хирургического лечения.
Заключение. Таким образом, приведенные в настоящей статье клинические наблюдения хирургического лечения редкой врожденной патологии грудной клетки представляют клинический интерес и заслуживают внимания. Правильное понимание сути патологического процесса, лежащего в основе развития комбинированной ДГК, позволяет использовать адекватное хирургическое пособие. Представления о высоком риске осложнений и травматичности операции у этой группы пациентов преувеличены.
Это заболевание впервые было описано J. Martin, J. Bell в 1943 г. В 1969 г. H. Lubs обнаружил хромосомный маркер — хромосому X с пробелом в субтеломерном участке длинного плеча Xq27.3. Отсюда основное название синдрома — синдром фрагильной (ломкой) Х-хромосомы. В 1991 г. удалось показать, что при этом синдроме множественные повторы последовательности CGG в Xq27.3 являются причиной локального гиперметилирования и повреждения синтеза белка [Verk ег К. et al., 1991]. В общей популяции здоровые индивидуумы имеют от 5 до 50 таких тринуклеотидных повторов, носители же мутантного гена FMR1 — от 50 до 200 повторов. Если же число повторов превышает 200, то появляется полный фенотип синдрома ломкой хромосомы X, и метилированный FMR1 ген не продуцирует белок [Devys D. e t al., 1993]. Функции белка FMRP неизвестны. Предполагается, что его отсутствие сказывается на процессах развития ЦНС. В мозге этот белок присутствует во всех нейронах и более представлен в сером веществе. В период эмбрионального развития концентрация FMRP особенно велика в базальном гигантоклеточном ядре, которое является поставщиком холинергических нейронов для лимбической системы.
Существует широкий спектр нарушений у больных с синдромом ломкой Х-хромосомы. Лица женского пола с полной мутацией более сохранны, чем лица мужского пола. У них в 30 % случаев не отмечается умственной отсталости.
Частота встречаемости 1:2000 у лиц мужского пола и от 2,5 до 6 случаев на 100 детей с УМО [Herbst D. S., Miller J. R., 1980; Hagerman R. et al., 1988].
Патогенез заболевания остается невыясненным.
Больным с Х-ФРА свойствен специфический физический фенотип, определяемый следующими стигмами дизонтогенеза. Дети имеют череп долихоцефалической формы, удлиненное лицо с выступающим лбом и прогенией. Ушные раковины оттопырены и увеличены. Нос с широким основанием, кончик его клювовидный, опущены углы рта, нередко встречаются высокое небо, подслизистые расщелины неба и язычка. Средняя часть лица уплощена. Пальцы рук удлинены, стопы плоские. Наблюдаются макроорхизм (после пубертатного периода), гипотония.
Повышена эластичность кожных покровов, аорты, сердечных клапанов. Отмечается слабость связочного аппарата коленных и голеностопных суставов [Козлова С. И. и др., 1987; Маринчева Г. С. и др., 1988; Денисова Л. В., 1988; Gillberg Ch. , 1995].
Наблюдается отставание в умственном и речевом развитии. Когнитивные функции недостаточны; IQ варьирует от 70 до 35, в большинстве случаев ниже 50 [Hagerman R. J., Suverman A., 1991]. Речь с небольшим запасом слов. Часты и более высокие достижения в вербальных задачах. Большинство девочек имеют IQ от нормального уровня и ниже нормы — 80—90. Отмечаются дизлексия, дискалькулия.
У ряда больных выявляются аутистические расстройства. В первые месяцы жизни дети развиваются нормально, в редких случаях отстает становление крупных моторных актов, наблюдается мышечная гипотония с тенденцией к прогрессированию. Иногда отмечаются беспокойство и плач с первого месяца жизни. После года — полутора лет становится заметным отставание в умственном развитии, замедляются формирование речи, пополнение словарного запаса, длительно сохраняется одно- или двусложная фраза. Дети на этом этапе привязаны к матери, мало стремятся к общению со сверстниками. Ограниченность в общении постепенно сочетается с появлением отторжения тактильного контакта с матерью, робостью с избеганием взгляда, задержкой формирования глазной реакции, слежения. Со времени становления ходьбы обнаруживается двигательная расторможенность. К 3—4 годам идет становление мелкой моторики рук, при этом заметно отстает формирование навыков самообслуживания, моторные акты примитивны, обеднены. Тогда же, иногда с начала 2-го года, появляются манерные движения в пальцах рук, отдаленно напоминающие «ручные манеризмы в пальцах и кистях рук» у детей с синдромом Каннера. Игра сохраняет примитивный, повторяющийся характер, протекает в одиночестве.
Бедность словарного запаса с годами становится очевидной, появляются неравномерность темпа речи, однообразность тембра, неуправляемая громкость. Речевой поток с ускоренным выпаливанием отдельных слов сменяется затуханием громкости речи с возникновением нечеткости в произношении звуков, что создает похожесть ее на эгоцентрическую речь у детей с детским аутизмом. При этом само общение нарушается и начинает походить на аутистическое с отказом от социальных контактов со сверстниками и родными.
К особенностям отрешенного поведения у этих детей следует отнести неровный осциллирующий характер отрешенности (на протяжении коротких отрезков времени), периодическую замену ее активным стремлением ребенка к более полноценному общению. В периоды подъема активности уменьшается робость, появляется тенденция к глазной реакции, тактильному и речевому контакту, исчезает дефицит внимания, на недлительный срок как бы смягчается аутизм. В последующем вновь возвращаются аутистические черты в поведении. Их неустойчивость и неполная выраженность (в сравнении с классическим аутизмом) постоянно сохраняются. В периоды спадов активности, свойственных этим детям, наблюдается переход от более высоких форм реагирования к более упрощенным, выявляются черты более раннего примитивного поведения, стереотипии в моторной и речевой сферах, вплоть до полного угнетения активности. Исчезает глазная реакция, возникает «взгляд в никуда». Пропадают ответы на обращенную речь. Появляются речь с самим собой, однообразные вращательные движения кистями рук, повороты вокруг собственной оси.
Наличие в клинической картине больных с Х-ФРА таких переходов в активности и реагировании только отдаленно напоминает симптом переслаивания функций у больных с синдромом аутизма Каннера, но не идентично ему. У больных с Х-ФРА нет истинного смешения ранних и более поздних функций, а наблюдается более целостное реагирование, как бы соответствующее то более раннему, то более зрелому возрасту. Дезинтеграции в разных функциональных системах не возникает. Следует отметить, что имеются единичные случаи с Х-ФРА, неотличимые от синдрома раннего детского аутизма.
На последующих возрастных этапах в личностной структуре больных с Х-ФРА сохраняются черты сенситивности, повышенной чувствительности, смущаемости с быстрым отказом от любого общения, с избеганием глазного контакта. Возрастает количество привычных моторных стереотипии в виде потирания ладоней рук, потряхивания кистями рук. В речи отмечаются стереотипные повторы слов, эхолалии слов, фраз. На фоне появляющегося интереса к окружающим детям контакты с ними практически не формируются, общение затруднено, но не достигает глубины классического аутизма.
С годами деятельность становится монотоннее, все более упрощаются интересы, побуждения становятся резко недостаточными. Больные обращаются к одним и тем же игровым сюжетам, игровые фантазии отличаются крайней обедненностью сюжетов, из года в год поведение повторяется, как клише. Углубляются трудности перехода к новым формам деятельности. Поведение становится примитивным, в нем нет парадоксальности и вычурности.
Эмоциональное развитие, привязанность к родным соответствуют уровню психического развития.
Наличие примитивных моторных стереотипии и бедность побудительных мотивов являются помехой в усложнении и формировании моторных навыков, необходимых в самообслуживании. Особое внимание обращает на себя нарастающая торпидность в мышлении, действиях, поведении. При этом легко возникают реакции раздражительности, протеста, а также невротические реакции в ответ на психогенные воздействия.
С увеличением возраста больных все грубее становятся когнитивные проблемы. Уровень IQ не возрастает, умственная отсталость (без диссоциированности) не смягчается, а достигает устойчивой стабилизации. Структура интеллектуального дефекта носит равномерный характер.
По данным R. Hodapp и соавт. (1990), в пубертате возможна остановка в развитии, с регрессом и значительным снижением IQ, с сохранением робости, избегания взгляда.
Свойственные этим детям спады и подъемы активности смягчаются. Аутизм проявляется в сужении круга общения, стереотипной, обедненной деятельности, в недостаточности речевого общения. Уровень социализации соответствует тяжести умственного недоразвития, в большинстве своем эти дети нуждаются в уходе и надзоре в течение всего периода жизни, многие дети заканчивают свою жизнь в учреждениях собеса.
При проведении дифференциального диагноза с аутизмом шизофренического спектра следует опираться на отсутствие диссоциации в умственном развитии, особых манеризмов в моторике, смягчение аутизма с возрастом ребенка, частично на фоне лечения и реабилитации.
Дифференциальный диагноз помогает проводить цитогенетический анализ, показывающий Х-ломкую хромосому в 2—70 % случаев. Аутизм при Х-ФРА в ряде случаев носит нерезко выраженный характер и не ведет к дезинтеграции в деятельности.
Терапия. Считается, что специфического лечения при Х-ФРА нет [Hagerman R., Silverman A., 1991]. Многие авторы предлагают пользоваться стимуляторами [Hagemian R. J., M urphy M. A., Wittenberger M. D., 1988].
Для уточнения диагноза необходимо проводить исследование на молекулярном уровне.
Таким образом, аутистический круг расстройств при Х-ФРА возникает позже, после 1—2 лет жизни ребенка; глубина его меньше, чем при классическом аутизме, моторные стереотипии проще и охватывают более зрелые моторные формулы, эмоциональная сфера никогда не бывает такой скудной, глазная реакция более сформирована или вовсе не повреждена, нет синдрома переслаивания зрелых и менее зрелых невытесненных функций. Все перечисленные особенности аутистическиподобных расстройств при Х-ФРА и послужили основанием к тому, чтобы их определять аутистическиподобными, а не аутистическими симптомами.
Однако и аутистическиподобный комплекс симптомов при Х-ФРА несет в себе основные признаки аутизма: отрешенность, стереотипность в деятельности и движениях, диссоциацию в развитии. Возможно, что аутистическиподобные синдромы при Х-ФРА связаны с поражением не вполне идентичных с синдромом Каннера структур мозга, но, по-видимому, все же очень близких и определенным образом взаимосвязанных, что подлежит дальнейшему изучению.
Приводим клиническое наблюдение больного с Х-ФРА
Ребенок М., 9 лет, от 1-й беременности, первых родов. Раннее психомоторное развитие близко к возрастной норме. После полугода стал отставать в становлении речи, крупных моторных актов. С годами нарастали возбудимость, раздражительность, моторное беспокойство. С детьми не играл. В обособленной игре (в одиночку) игрушки обнюхивал, вертел, лизал. Тактильный контакт с матерью отвергал. Настроение было снижено, часто без внешних на то причин кричал, бил себя, появлялась тенденция к разрушительству. С годами возрастала неуправляемость.
Психический статус при поступлении. Общение отвергает. Негативен. Осмотру подчиняется с трудом. На вопросы отвечает невнятно. Речь — одно-, двусловная, с аграмматизмами. Речевая тональность, тембр изменены. Словарный запас обеднен. Речь легко становится невнятной, затухшей, близкой к эгоцентрической. Периодами истощается и уходит от общения, в беспокойстве перемещается по кабинету, оставляя без внимания все попытки общения с ним. Через несколько минут вновь удается на короткий срок привлечь его внимание. Энергетический потенциал флюктуирующий и очень низкий, любая деятельность вначале отвергается ребенком, после активного многократного побуждения к ней удается получить один-два ответа в плане вопросов, а затем вновь наступают отторжение контакта, моторное беспокойство с раздражительностью. В кистях рук сохраняются примитивные атетозоподобные стереотипии. В целом поведение полевое, с временами наступающим возбуждением, самоагрессией и разрушительными тенденциями.
В психическом статусе на первый план выступает тяжелое умственное недоразвитие с аутистическими тенденциями, которые позволили диагностировать детский аутизм. Структура аутизма была столь глубока и очевидна, что на этом этапе состояние приближалось к детскому аутизму Каннера.
Логопедическое обследование. Задержка психоречевого развития, аутистический синдром.
Неврологический статус. ЧМН — без видимой патологии. Ходит самостоятельно. Тонкая моторика кистей рук не сформирована. Сухожильные рефлексы не изменены. Координация не нарушена. Патологических знаков нет. Болевая чувствительность сохранена. Тазовые функции не нарушены.
Соматический статус. Долихоцефальный череп, удлиненное лицо, массивный подбородок. Большие оттопыренные уши, прогнатизм, высокое небо, нос с широким основанием и клювовидным кончиком. Кисти и стопы увеличены. Кожа гиперэластичная, растяжима. Суставы с повышенной разгибаемостью. Пролапс митрального клапана. Другой патологии со стороны внутренних органов не отмечено.
Катамнез: 9 лет 10 мес. После проведения 1-го курса лечения церебролизином по методу Осипенко — Скворцова смягчились моторная расторможенность и аутистическая отрешенность. Улучшились ориентация в окружающем, память, увеличился словарный запас, стал усваивать новые знания. В связи с заболеванием сестры проведено цитогенетическое обследование и кариотипирование детей и матери. Обнаружены 46 XY, Х-ФРА. У пробанда и сибса при цитогенетическом исследовании обнаружена ломкая Х-хромосома.
Катамнез: 10 лет 9 мес. После трех курсов терапии по методу Осипенко — Скворцова в НТЦ состояние резко улучшилось. Расширилось общение с окружающими, смягчилась аутизация. Появилось стремление к игровой деятельности, способен к наблюдению и подражанию в ней. Начал обучаться в школе. Выучил буквы. Сидит на уроках самостоятельно, без матери.
Полностью исчезли моторное возбуждение, беспричинный страх, бесцельное беспокойство кататоноподобного характера. В кистях рук не стало манеризмов. Осознает временные события. Стал пользоваться личными местоимениями, восстановилось осознание себя, использует личные местоимения по отношению к себе. Исчезло симбиотическое отношение к матери. Помогает ей в уходе за сестрой. Оживились эмоции.
Логопедическое обследование. Улучшилась связанная речь, стал употреблять более сложные по звуковому составу слова.
Заключение. В настоящее время в статусе на первый план выступает умственное недоразвитие со значительным нивелированием аутистической симптоматики, снятием кататоноподобного возбуждения и страхов. При первом осмотре ребенка в возрасте 9 лет статус определяли проявления аутизма, стереотипии в пальцах рук, симптомы тождества, особые речевые расстройства в форме контаминации, незавершенности фраз, их разорванности, негативизм, фобии, моторное возбуждение с агрессией, которые служили основанием для диагностики аутизма.
Периодами состояние утяжелялось, сопровождаясь регрессом приобретенных навыков, присоединением фобического синдрома, оживлением моторного возбуждения с агрессивными тенденциями. Наличие этих симптомов, казалось бы, только подтверждало диагностику классического аутизма.
Положительная динамика состояния с почти полным купированием аутистических расстройств, обнаружение при цитогенетическом обследовании ломкой Х-хромосомы послужили основанием к постановке диагноза: УМО, Х-ФРА, аутистическиподобный синдром.
Настоящий случай представляет несомненный интерес в отношении как динамики состояния при лечении новым методом по Осипенко — Скворцову, так и нивелирования собственно аутистических симптомов при Х-ФРА, а также уменьшения проявлений умственного недоразвития. Следующее наблюдение.
Ребенок М., 5 лет 1 мес, от 2-й беременности, протекавшей без патологии. Роды в срок, нормальные .
Психомоторное развитие. Голову держит с 1 мес, сидит с 6 мес, ходит с 13 мес. Первые слова с 1,5 года; фразовой речи нет.
К 3 годам в поведении стали обращать на себя внимание задержка в становлении речи, отказ от общения, однообразная стереотипная, примитивная игра, особые постукивания пальцами рук по предметам, сосание пальцев рук, периодические двигательные возбуждения с негативностью.
В 4,5 года поступила в НТЦ профилактики и лечения детской неврологической инвалидности.
Психический статус при поступлении. Отрешена. Глазной реакции нет, на звук, зов реакция отставленная. Обследованию сопротивляется. Речь эгоцентрическая, понимание элементарной фразы присутствует, ответы иногда носят противоположный характер. Настроение индифферентно-раздражительное. На показ игровых предметов, сладостей положительно не реагирует, напротив, следует отказная, протестная реакция, стремление к уходу от источника раздражения. В целом поведение однообразное, бездеятельное. В пальцах рук стереотипные манеризмы. Сон не расстроен, аппетит вялый, избирательный выбор пищи.
Неврологический статус. ЧМН без патологии. Двигательная сфера: ходит самостоятельно, расторможена. Сухожильные рефлексы живые, равномерные. Патологических знаков нет. Координация не нарушена. Болевая чувствительность сохранена. Дисфазия.
ЭЭГ: -ритм 7 кол/с, регулярный, 1000 мкВ. Эпикомплексы отсутствуют; -ритм незначительный, -волны — множественные.
Изменения на ЭЭГ резидуально-органического генеза свидетельствуют о дисфункции диэнцефально-стволовых структур.
Диагноз: атипичный РДА, сдвиг в развитии между 1 — 1,5—3 годами. Отставание в умственном развитии на 2— 3,5 возрастных порядка.
После двух курсов терапии в НТЦ состояние ребенка улучшилось. Увеличился словарный запас, стала иногда пользоваться двусловными предложениями. Речь не использует в качестве средства коммуникации. По инструкции не работает. Аутохтонная деятельность расширилась: реагирует на детские передачи, показанные по телевизору, иногда наблюдает за игрою детей. Стала периодами выполнять просьбы матери. Можно сказать, что отрешение стало менее выраженным, больше осознает ситуацию.
Катамнез: 5 лет 5 мес. После курсов терапии в НТЦ отмечена положительная динамика в состоянии ребенка. Улучшилось в целом самочувствие, нивелировалась депрессия, смягчилась астения. Настроение стало ровнее. Появилась ориентировочная реакция на зов, звук. Восстановилась глазная реакция с возможностью зрительной фиксации на предметах. Ребенок стал пользоваться лепетными формами речи, особенно в случаях аффективного напряжения. Дольше может смотреть телевизор. Стала подчиняться матери, появилась возможность вывести ее на прогулку. Увеличилась протяженность действий по заданию извне. При поступлении направленная деятельность отсутствовала. В моторике кистей рук сохраняются стереотипные манеризмы, присутствует ходьба на цыпочках наряду с обычной опорой во время ходьбы на всю стопу. Реакция «глаза в глаза» стала продолжительнее во времени. Негативистические, отказные реакции остаются, однако они наблюдаются реже.
ЭЭГ-изменения резидуально-органического генеза; в сравнении с предыдущей ЭЭГ установлено некоторое улучшение параметров корковой активности; -ритм — 8 кол/с.
Логопедическое обследование. Активизировался лепет, легче идет на занятия к логопеду.
Цитогенетическое исследование. При цитогенетическом исследовании и детей, и матери: обнаружена ломкость участка Xq27.3, позволившая подтвердить диагноз Х-ФРА.
Таким образом, на основании обследования и наблюдения можно подтвердить диагноз: синдром Мартина — Белл, или Х-сцепленная умственная отсталость с ломкой Х-хромосомой (Х-ФРА), аутистическиподобный синдром.
Интерес в данном случае представляет поздняя диагностика Х-ФРА, коморбидность с аутистическиподобным синдромом, большое сходство болезненного состояния с процессуальным аутизмом, значительное улучшение состояния после терапии.
Синдром Робинова
Синдром Робинова — это орфанное наследственное заболевание, которое характеризуется задержкой роста с множественными фенотипическими аномалиями. Патология возникает при аутосомно-рецессивном и аутосомно-доминантном наследовании мутаций. Болезнь характеризуется мезомелической карликовостью, патогномоничными врожденными пороками строения лица, отставанием в развитии. С диагностической целью проводятся специальные молекулярно-генетические исследования, применяются различные методы визуализации. При синдроме Робинова возможно только симптоматическое лечение: хирургическая коррекция аномалий развития, нейропсихологическая программа реабилитации больных.
МКБ-10
Общие сведения
Мезомелическая карликовость была описана в 1969 г. американским генетиком М. Робинов в сотрудничестве с врачами Ф. Сильверманом и Х. Смитом, поэтому в некоторых медицинских источниках болезнь называют синдромом Робинова-Сильвермана-Смита. Патология встречается крайне редко — 1 случай на 500 тыс. новорожденных. Наибольшая база данных о таких пациентах собрана в США благодаря Фонду синдрома Робинова, который занимается статистической работой, оказывает поддержку людям, столкнувшимся с этим заболеванием.
Причины
Благодаря достижениям современной генетики в 2000 г. были установлены точные генетические предпосылки синдрома Робинова. Заболевание может наследоваться по аутосомно-доминантному типу (когда болен один из родителей), по аутосомно-рецессивному типу (если оба родителя — носители мутантного гена). Развитие мезомелической карликовости вызвано следующими генными дефектами:
- МутацииWNT5A, DVL1, DVL3. Все три варианта генетических дефектов передаются аутосомно-доминантным путем. Такой тип синдрома Робинова отличается менее выраженными аномалиями развития, имеет благоприятный прогноз для больных.
- МутацииROR2. Патология этого гена наследуется аутосомно-рецессивным путем, вызывает грубые внутриутробные пороки и, кроме того, обычно сочетается с интеллектуальными расстройствами.
Главным предрасполагающим фактором развития синдрома Робинова являются близкородственные браки, при которых в популяции накапливается большое число мутаций, резко возрастает риск неблагоприятной комбинации генетического материала родителей. Поэтому максимальное распространение болезни наблюдается в Турции и Омане, где браки между родственниками до сих пор встречаются довольно часто. Влияние тератогенных факторов в эмбриональном периоде беременности не исключается, но точных научных данных по этому вопросу пока нет.
Патогенез
Характерные клинические проявления синдрома Робинова обусловлены нарушениями процессов созревания и дифференцировка тканей, которые возникают во время эмбрионального развития ребенка. При аутосомно-доминантном типе синдрома в период эмбриогенеза наблюдается недостаточность протеинов из группы WNT, которые необходимы для правильного формирования соединительной ткани и образования скелета.
Для аутосомно-рецессивного варианта болезни характерны мутации гена, отвечающего за работу тирозинкиназного рецептора, который расположен на поверхности всех типов клеток организма эмбриона. При недостаточности ROR2 происходят грубые нарушения роста и размножения клеток, изменяются биохимические реакции клеточного метаболизма. Вследствие таких мутаций страдает костная и хрящевая ткань, а также ткани внутренних органов.
Симптомы
Признаки патологии четко видны сразу после родов. Особенности черепно-лицевого строения включают большой размер головы (макроцефалию), крупные выпуклые глаза, короткий вздернутый нос с расширенными ноздрями и вдавленной переносицей. Также у ребенка заметны большой рот (макростомия), обнаженные передние зубы в спокойном состоянии, недоразвитая нижняя челюсть (микрогнатия). Зачастую наблюдается выпуклый высокий лоб, низко расположенные уши.
Типичные нарушения строения тела заключаются в мезомелии — недоразвитии средних участков верхних и нижних конечностей (предплечья, голени). Аномалии развития скелета дополняются брахидактилией (короткопалостью), врожденным сколиозом, который вызван пороками строения грудных и шейных позвонков. По мере взросления ребенка становится заменой задержка роста, избыточный вес.
Синдром Робинова, как правило, сопровождается нарушениями полового развития: у девочек выявляется недоразвитие клитора и больших половых губ, у мальчиков — тяжелая гипоплазия полового члена. Если заболевание связано с аутосомно-доминантным типом мутации, клинические симптомы выражены не так интенсивно, отсутствует патологическая полнота, а половое развитие находится в пределах нормы.
Осложнения
Синдрому Робинова нередко сопутствуют соматические болезни, вызванные нарушениями внутриутробного развития. К типичным осложнениям относят гидронефроз с последующей атрофией органа, аномалии формы, размера или положения почек. Нередко диагностируются врожденные паховые или пупочные грыжи. У 10% больных развиваются врожденные пороки сердца и крупных сосудов.
Около 20% пациентов с синдромом Робинова имеют нарушения психического развития по типу умственной отсталости легкой или среднетяжелой степени. В сочетании с выраженными аномалиями скелета это нарушает социализацию детей, провоцирует серьезные психологические проблемы. Вследствие скелетных аномалий такие больные неспособны к большинству видов трудовой деятельности, потому они нуждаются в медико-социальной помощи.
Диагностика
Зачастую фенотипические признаки синдрома Робинова определяются еще в периоде внутриутробного развития при плановом ультразвуковом скрининге. Однако исследование не дает 100% вероятности диагноза, поэтому обследование продолжается после рождения ребенка. Чтобы установить мезомелическую карликовость, врачу требуется полное физикальное обследование пациента и следующие инструментально-лабораторные методы:
- Рентгенография скелета. Рентгенологическое исследование демонстрирует характерное мезомелическое укорочение конечностей, разнообразные аномалии лицевого черепа и другие отклонения от физиологической нормы.
- УЗИ сердца. Учитывая высокую частоту сердечных пороков у страдающих синдромом Робинова, ультразвуковое сканирование и консультация кардиолога (если это необходимо) являются важным этапом диагностики.
- УЗИ органов живота. Инструментальная визуализация используется для оценки структурно-функциональных особенностей почек, мочевыводящих путей, подтверждения диагноза при обнаружении грыж передней брюшной стенки.
- Генетическое тестирование.Молекулярно-генетический анализ методом автоматического секвенирования проводится для диагностики болезни Робинова, выяснения ее клинического варианта, что важно для оценки прогноза заболевания.
Лечение синдрома Робинова
Специфические методы терапии отсутствуют. Медикаментозная терапия при этом заболевании неэффективна, однако она может применяться при сопутствующих соматических аномалиях для стабилизации состояния пациента. Основную роль в лечении таких больных играет помощь пластических и челюстно-лицевых хирургов для максимально возможного устранения скелетных аномалий. Целесообразно проведение следующих типов операций:
- экстракраниальная или интракраниальная коррекция гипертелоризма;
- коррекция положения нижней челюсти с ее фиксацией пластинами и установкой имплантатов для добавления недостающего объема;
- стоматологические операции для восстановления нормального прикуса;
- коррекция длины конечностей с помощью хирургической реконструкции.
Операции выполняются, начиная с раннего возраста ребенка, причем у каждого типа вмешательства есть собственные временные рамки и показания. Для максимально полного устранения недостатков внешности пациентам требуются сложные многоэтапные вмешательства. При наличии жизнеугрожающих пороков хирургическое лечение рекомендовано начать в ранние сроки, чтобы восстановить функционирование организма.
Если синдром Робинова сопровождается интеллектуальными нарушениями, больным требуется помощь педагогов-дефектологов. При легкой степени расстройств дети успешно обучаются по специальным программам, сохраняют работоспособность. При среднетяжелой выраженности нарушений речевое и психическое развитие значительно снижены, поэтому суть реабилитации заключается в привитии пациенту навыков самообслуживания.
Прогноз и профилактика
Синдром Робинова существенно не влияет на продолжительность жизни, но значительно ухудшает ее качество, особенно при несвоевременном проведении пластических операций по коррекции скелетных аномалий. При многоэтапной хирургической помощи удается достичь естественной внешности, улучшить функциональные возможности скелета. У пациентов, страдающих мезомелической карликовостью и сопутствующими соматическими пороками, прогноз неблагоприятен.
Если в семье были случаи диагностированного синдрома Робинова или карликовости, вероятно связанной с генетическими патологиями, перед планированием ребенка необходима консультация генетика. При обследовании обоих партнеров проводится анализ на типичные мутации, на основании результатов которых устанавливается риск рождения больного ребенка. В дальнейшем такие пары подлежат генетическому консультированию на протяжении беременности.
2. WNT5A mutations in patients with autosomal dominant Robinow syndrome/ Person A. D., Beiraghi S., Sieben C. M., Hermanson S., Neumann A. N., Robu M. E., Schleiffarth J. R., Billington C. J. et al// Dev Dyn. — 2010.
3. Клинический случая синдрома Робинова в сочетании с множественным гамартозным ростом/ Ю.Б. Гречанина, Л.В. Молодан. — 2003.
Читайте также: