Синдром Страйкера-Халбейзена (Stryker-Halbeisen) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 22.01.2026
ГБОУ ВПО «Московский государственный медико-стоматологический университет им. А.И. Евдокимова» Минздрава России, Российская Федерация
Стоматологический факультет, кафедра детской терапевтической стоматологии
Особенности лечения детей с синдромом Халлермана—Штрайфа на стоматологическом приеме
Мандибулоокулофациальный синдром (Нallerman—Streiff syndrome) — это наследственное спорадическое заболевание, характеризующееся низким ростом пациента с пропорциональным телосложением и характерным «птичьим лицом». Соотношение полов — 1:1. Популяционная частота: от 1:10 000 до 1:30 000.
Синдром Халлермана—Штрайфа впервые был описан С. Audru в 1893 г. В 1948 г. немецкий офтальмолог W. Нallerman выделил его самостоятельную нозологическую форму, позднее швейцарский офтальмолог E. Streiff также описал клинические случаи проявления данной патологии.
Основные признаки: брахицефалия, выступающие теменные и лобные бугры, тонкий клювовидный нос, маленький рот, малые размеры верхней челюсти; гипоплазия хрящей носа и нижней челюсти и узкие верхние дыхательные пути обусловливают частые осложнения: затрудненное дыхание, обструктивное апноэ во время сна, рецидивирующие инфекции дыхательных путей. Наблюдается также двусторонняя катаракта, микрофтальмия; высокое небо; неправильный рост зубов и сверхкомплектные зубы; гипотрихоз вплоть до гнездной алопеции; очаговая атрофия кожи головы и носа. Интеллектуальное развитие, как правило, в пределах низкой возрастной нормы. Тип наследования не установлен. Риск повторения аномалии у детей пробанда: низкий. Пренатальная УЗ-диагностика обязательна. Лечение симптоматическое.
Родители восьмилетнего пациента В. обратились в детскую клинику МГМСУ им. А.И. Евдокимова для обследования и лечения по поводу нарушения прикуса у ребенка, изменения внешнего вида формы зубов, разрушения их коронок.
Анамнез соматического заболевания. Патология выявлена при рождении ребенка. Ребенок от второй беременности, вторых родов в 39 нед. Настоящая беременность со слов матери протекала без осложнений, без угрозы прерывания беременности. Вес при рождении 3700 г, рост 54 см, головное предлежание, закричал сразу. Оценка по шкале Апгар 7/8 баллов. На 2-е сутки в связи со стридором, невозможностью сосания переведен в отделение интенсивной терапии патологии новорожденных.
Зондовое вскармливание в течение 3 мес, далее до года только с ложки жидкой пищей. Самостоятельный прием пищи после года. Задержка физического, психомоторного и речевого развития: гипотрофия, голову держит с 3 мес, сидит с 9 мес, ходит с года. Лепетная речь с 2 лет. Фразовая речь с 3 лет (речь малопонятна, запас слов и речевых навыков ограничен).
Перенесенные заболевания: ОРВИ, ветряная оспа.
Диагноз: мандибулоокулофациальный синдром (синдром Халлермана—Штрайфа).
В возрасте 1 года при оформлении статуса инвалидности и диспансерным обследованием специалистами установлены следующие множественные врожденные пороки развития (МВПР). Сердечно-сосудистая система: врожденная патология — открытое овальное окно без нарушения кровообращения и регургитации, МАРС (дополнительные хорды левого желудочка), пролапс митрального клапана. Центральная нервная система: ишемически-гипоксически-энцефалопатический синдром, задержка физического, психомоторного и речевого развития. Брахицефалическая форма черепа, позднее закрытие родничков (большой родничок не закрыт). Патология глаз: микрофтальм, врожденный птоз верхних век, косоглазие; частичная атрофия зрительного нерва; астигматизм, миопия средней степени; обратное зрение, голубые склеры. Опорно-двигательный аппарат: низкий рост, кифосколиоз; узкая грудная клетка с деформацией ребер и мечевидного отростка; деформация ключиц и лопаток, костей и мышц правого предплечья. Гипогенитализм.
В настоящее время у ребенка низкий рост (97 см), несоответствие паспортного и физиологического возраста ребенка. Кожные покровы: гипотрихоз — волосы светлые, сухие, ломкие с участками алопеции; кожа светлая, сухая, тонкая с участками гиперпигментации, морщинистая.
При обследовании головы и полости рта выявлена следующая патология развития: дисцефалия с гипоплазией нижней челюсти и хрящей носа («птичье лицо»), врожденное недоразвитие тела и ветви нижней челюсти, дистальное положение нижней челюсти, микростомия. Деформация прикуса, дистальная окклюзия (смыкание по II классу Энгля), вертикальная резцовая дизокклюзия, сужение верхней челюсти, готическое небо и истинная микрогнатия и микрогения.
Задержка прорезывания постоянных зубов — сохранены все временные зубы, отсутствует их физиологическая подвижность, скученность зубов верхней и нижней челюсти, отсутствуют тремы. Эмаль зубов желтоватого цвета, матовая, не имеет блеска. Определяются участки истончения эмали и обнажения дентина. Зубы 5.4, 6.4 — коронки разрушены, дентин пигментированный, податливый. При зондировании отмечается болезненность (К04.3 хронический пульпит). Зубы 5.2, 5.1, 6.1, 6.2 — кариозные полости, заполненные размягченным пигментированным дентином на контактных и оральных поверхностях, зондирование болезненно (К04.3 хронический пульпит). 7.4, 8.4 — кариозные полости на жевательных поверхностях заполнены плотным пигментированным дентином (К02.1 кариес дентина). Гипоплазия эмали временных зубов верхней и нижней челюстей. Стираемость выражена незначительно.
Проведена дифференциальная диагностика со следующими заболеваниями: прогерия, мандибулофациальный дизостоз, синдром Секкеля.
На основании клинического, клинико-генеалогического, рентгенологического и специальных методов обследования поставлен диагноз: мандибулоокулофациальный синдром (синдром Халлермана—Штрайфа). Тип наследования не установлен.
На ортопантомограмме выявлено: недоразвитие нижней и верхней челюстей, скученность зубов, системная гипоплазия эмали, задержка сроков формирования и прорезывания зубов.
Проведена спиральная компьютерная томография лицевого отдела черепа, построена стереолитографическая модель (3D) черепа с последующим планированием проведения реконструкции нижней челюсти и скулоорбитального комплекса методом КДА и контурной пластики (в 12—14 лет: после окончания формирования и развития лицевого отдела черепа, соответствия паспортного и физического возраста). Комплексное ортодонтическое лечение на съемной и несъемной технике до и после остеотомии верхней челюсти и нижней челюсти с постановкой в конструктивный прикус (в 14—18 лет).
Проведена профессиональная гигиена полости рта, санация полости рта. Рекомендована реминерализующая терапия с целью профилактики кариеса и его осложнений. В дальнейшем рекомендовано диспансерное наблюдение у врача-стоматолога с кратностью осмотров 3—4 раза в год, проведение хирургической и ортодонтической коррекции соответственно возрасту пациента.
Необходима диспансеризация и комплексное лечение у детских врачей-специалистов — педиатра, кардиолога, офтальмолога, ортопеда, детского невролога, дерматолога, психолога.
Прогноз для жизни пациента с учетом обследования и лечения — благоприятный.
Синдром Штрикера-Хальбейзена (syndroma Stryker — Halbeisen)
Этот синдром встречается среди недостаточно питающихся людей. На коже лица, шеи и туловища появляется полиморфная сыпь, состоящая из сильно зудящих эритематозных пятен, пузырьков и мокнущих участков, и напоминающая собой острый или подострый дерматит, вызванный экзогенными факторами. В редких случаях этот дерматит протекает под видом десквамативной эритродермии. Со стороны крови отмечается макроцитоз с анемией.
Отличить эти кожные явления от экзогенного или медикаментозного дерматита, экземы, невродермита, некоторых вторичных или первичных эритродермий без данных исследования крови очень трудно.
Лечение, проводимое при соблюдении полноценного режима питания витамином В12, препаратами железа, экстрактом печени, пиридоксином, дает успешные результаты, что в известной мере подтверждает диагноз.
Тромбастения Гланцмана (thrombasthenia Glanzmann)
Это заболевание соответствует врожденной псевдогемофилии Francka и представляет собой наследственный геморрагический диатез, передающийся матерью чаще всего девочкам (в отличие от гемофилии).
Признаки характеризуются появлением самопроизвольных или вызванных травмами кровоизлияний на коже и из слизистых оболочек. Время кровотечения и свертывания крови при этом не изменяется. Количество тромбоцитов также остается в норме, но наступают значительные изменения их в качественном отношении, заключающиеся в дегенерации, базофильности, анизоцитозе, появлении гигантских форм, отсутствии способности агглютинировать и др.
Это заболевание сходно с тромбоцитолитической спленогенной пурпурой Kaznelsona, выражающейся кровоизлияниями на коже, из слизистых оболочек, почек и др. в результате дегенерации и гипоплазии тромбоцитов.
Геморрагические синдромы Glanzmanna и Kaznelsona следует отличать от гемофилии, геморрагических пурпур Werlhofa и Francka и от псевдогемофилии.
« Синтетическая дерматология»,
Любен Попов
Читайте далее:
Синдром Мэя-Тернера в практике рентгенэндоваскулярного хирурга

Александр Быстренков:
Три года назад мы занялись проблемами вен малого таза и их ролью в развитии эректильной дисфункции. В частности, выполняем операции по эмболизации вен простатического сплетения. В 2019 году участвовали в 15-м конгрессе «Мужское здоровье» в Сочи, где познакомились со специалистами из Москвы, Санкт-Петербурга, которые занимаются вопросами веногенной эректильной дисфункции, начали сотрудничать. К нам в клинику периодически поступают такие пациенты из России, и мы успешно решаем проблему эндоваскулярным способом. К сожалению, в Беларуси такие вмешательства практически не выполняются. Мы освоили методику одними из первых, интересовались наработками российских коллег.
Наблюдая пациентов с венозной утечкой, специалисты пришли к выводу, что при данной патологии часто имеет место артериовенозный конфликт — сдавление вен артериальными стволами в виде синдрома Мэя — Тернера. На этом фоне развивается варикозное расширение нижележащих вен. Другой вид артериовенозного конфликта — аорто-мезентериальный пинцет (в зарубежной литературе можно встретить под названием синдром орехокола или синдром Щелкунчика) — одна из частых причин варикоцеле.
Александр Быстренков:
Предварительно молодой человек обследовался в клинической больнице Святителя Луки (Санкт-Петербург) и отправил в ГОКБ результаты КТ, МРТ, необходимые для подбора стента. В этом, к слову, заключается одна из сложностей.
Представьте структуру в виде окружности. Если ее сдавливать, она превращается в овальную, затем практически в плоскую. И из исходной окружности диаметром 15-16 миллиметров можем получить овальную структуру 4 на 25-27 миллиметров. Кроме того, картина ангиографии отличается от картины КТ. Во время вмешательства у нас возникли вопросы по протяженности сдавления. За помощью обратились к российским коллегам.
Чем меньшую зону покрывает стент, тем лучше. Неправильный размер конструкции увеличивает риск тромбоза. Стент — это в любом случае инородное тело, и необходимо время, чтобы он вжился в стенку сосуда. В случае с нашим пациентом зона сдавления оказалась небольшой.
Процедура имплантации стента заняла около часа, выполнена под местной анестезией в зоне прокола вены. Пациента выписали через сутки.
В Беларуси достаточно много вмешательств проводятся эндоваскулярно, например, стентирование коронарных, периферических, сонных артерий. Большинство таких процедур выполняется под местной анестезией. Необходимо обезболить точку пункции, чаще бедренной артерии, вены. На внутренних стенках сосудов отсутствуют болевые рецепторы.
Пациент отмечает: тазовые боли уменьшились. Однако венозная утечка — причина эректильной дисфункции — сохраняется, так как сохраняется варикозное расширение вен. Проблему врачи устранят на втором этапе — эмболизации простатического венозного сплетения.
Сложности диагностики
Александр Быстренков отмечает:
Синдром Мэя — Тернера не редкость. Однако это коварная патология, которая сложно диагностируется.
Пациенты жалуются урологу на боли в промежности при физической нагрузке, после полового акта, могут выявляться изменения на спермограмме, иногда общего анализа мочи, простатического секрета. Кроме того, заболеванию часто сопутствует варикоцеле, варикозное расширение вен нижних конечностей. Симптоматика, без учета патологии вен, хорошо укладывается в клинику хронического простатита. Пациенты подвергаются длительному лечению, и оно малоэффективно, так как причина в другом.
Синдром Мэя — Тернера представляет собой совокупность двух факторов. Во-первых, на фоне повышенного венозного давления в малом тазу развивается синдром хронических тазовых болей. Во-вторых, вследствие варикозного расширения вен появляется венозная утечка полового члена и, как результат, эректильная дисфункция. Сдавление левой общей подвздошной вены — широко распространенная патология, которая, по разным данным, встречается у 22-50 % населения.
Распространенность же клинически манифестированных форм артериовенозных конфликтов оценить сложно, так как это пациенты с весьма широким спектром проявлений: варикоцеле, эректильная дисфункция, синдром хронических тазовых болей, илеофеморальные тромбозы и посттромбофлебитический синдром, варикозное расширение вен малого таза, наружных половых органов.
На УЗИ взаимоотношения вен таза, почек, нижней полой вены оценить сложно. Но назначать пациенту с хроническим простатитом КТ или МРТ венозного русла малого таза, включая нижнюю полую и почечные вены, не всегда целесообразно. Поэтому нужно искать подсказки среди других маркеров — предрасположенность к варикозу, варикоцеле в анамнезе, характеристики тазовых болей и их взаимосвязь с возможным венозным полнокровием после полового акта и физических нагрузок.
Проблема синдрома Мэя — Тернера заключается и в том, что сдавление подвздошной вены часто приводит к образованию тромбов. По данным мировых исследований, отмечает Александр Быстренков, с левой стороны тромбоз подвздошной вены случается в 5 раз чаще.
Синдром Мэя — Тернера изучают в медицинских университетах. Однако на практике патологии не уделяют должного внимания, считая, что это нечто крайне редкое. Отсутствует настороженность в отношении данного заболевания, нет междисциплинарного взаимодействия. Специалисты назначают УЗИ, диагностические тесты, но не всегда обращают внимание на систему вен малого таза, почечные и нижнюю полую вены. При этом диагностика и лечение выполняются в рамках протоколов и стандартов, того, чему нас обучали в университете, на курсах повышения квалификации. Мы первые в стране провели такую операцию, а проблема давняя.
Синдром Мэя — Тернера — острый вопрос во всем мире. Операции по устранению патологии выполняются в западных странах, однако сказать, что налажена четкая система диагностики и лечения, нельзя.
В России немногие специалисты занимаются подобными проблемами, поэтому пациенты приезжают и к нам.
Работает команда
Операционная бригада состояла из двух рентгенэндоваскулярных хирургов. Это Александр Быстренков и заведующий рентгеноперационной Гомельского областного клинического кардиологического центра, главный внештатный рентгенэндоваскулярный хирург ГУЗО Гомельского облисполкома Сергей Гороховский.
Также в установке стента участвовали заведующий отделением сосудистой хирургии ГОККЦ, главный внештатный ангиохирург ГУЗО Гомельского облисполкома, кандидат мед. наук Алексей Печенкин, ассистент кафедры хирургических болезней № 3 ГомГМУ Александр Шестерня.
Периоперационным ведением, координацией обследования пациента занимались врач-хирург-уролог хирургического отделения (трансплантации, реконструктивной и эндокринной хирургии) РНПЦ радиационной медицины и экологии человека, кандидат мед. наук Эдуард Повелица и доцент кафедры урологии и нефрологии БелМАПО, кандидат мед. наук Николай Доста.
Во время вмешательства хирургам помогали операционная медсестра рентгеновского отделения Любовь Резниченко и рентгенолаборант Елена Добросельская.
Республиканское унитарное предприятие
«Редакция газеты «Медицинский вестник»
Болезнь Эрдгейма-Честера Обзор литературы и клинический случай
Ключевые слова
Об авторах
ФГБУ «Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина» Минздрава России
Россия
Александр Сергеевич Крылов
115478 Москва, Каширское шоссе, 24
Список литературы
1. Estrada-Veras J.I., O’Brien K.J., Boyd L.C. et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv 2017;1(6):357-66. DOI: 10.1182/bloodadvances.2016001784.
2. Emile J.F., Abla O., Fraitag S. et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127(22):2672-81. DOI: 10.1182/blood-2016-01-690636.
3. Arnaud L., Gorochov G., Charlotte F. et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood 2011;117(10):2783-90. DOI: 10.1182/blood-2010-10-313510.
4. Tran T.A., Fabre M., Pariente D. et al. Erdheim-Chester disease in childhood: a challenging diagnosis and treatment. J Pediatr Hematol Oncol 2009;31(10):782-6. DOI: 10.1097/MPH.0b013e3181b76827.
6. Campo E., Harris N.L., Jaffe E.S. et al. WHO Classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer 2017. P. 586.
7. Emile J.F., Charlotte F., Amoura Z., Haroche J. BRAF mutations in Erdheim- Chester disease. J Clin Oncol 2013;31(3):398. DOI: 10.1200/JCO.2012.46.9676.
8. Cangi M.G., Biavasco R., Cavalli G. et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis 2015;74(8):1596-602. DOI: 10.1136/annrheumdis-2013-204924.
9. Emile J.F., Diamond E.L., Hélias-Rodzewicz Z. et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood 2014;124(19):3016-9. DOI: 10.1182/blood-2014-04-570937.
10. Diamond E.L., Abdel-Wahab O., Pentsova E. et al. Detection of an NRAS mutation in Erdheim-Chester disease. Blood 2013;122(6):1089-91. DOI: 10.1182/blood-2013-02-482984.
11. Chester W. Uber lipoid granulomatose. Virchows Arch (Pathol Anat Phys) 1930;279:561-602.
13. Haroche J., Arnaud L., Amoura Z. Erdheim-Chester disease. Curr Opin Rheumatol 2012;24(1):53-9. DOI: 10.1097/BOR.0b013e32834d861d.
16. Scolaro J.C., Peiris A.N. The Hairy Kidney of Erdheim-Chester Disease. Mayo Clin Proc 2018;93(5):671. DOI: 10.1016/j.mayocp.2018.03.003.
17. Kraniotis P., Daoussis D. Periaortitis, hairy kidneys and bone lesions. Rheumatology (Oxford) 2016;55(12):2118. DOI: 10.1093/rheumatology/kew331.
18. Lee H.J., Lee K.Y., Shin D.Y. et al. A case of Erdheim-Chester disease with asymptomatic renal involvement. Cancer Res Treat 2012;44(2):146-50. DOI: 10.4143/crt.2012.44.2.146.
19. Serratrice J., Granel B., De Roux C. et al. “Coated aorta”: a new sign of Erdheim- Chester disease. J Rheumatol 2000;27(6):1550-3.
21. Suzuki H., Wanibuchi M., Komatsu K. et al. Erdheim-Chester disease involving the central nervous system with the unique appearance of a coated vertebral artery. NMC Case Rep J 2016;3(4):125-8. DOI: 10.2176/nmccrj.cr.2015-0331.
22. Diamond E.L., Hatzoglou V., Patel S. et al. Diffuse reduction of cerebral grey matter volumes in Erdheim-Chester disease. Orphanet J Rare Dis 2016;11(1): 109. DOI: 10.1186/s13023-016-0490-3.
23. Lachenal F., Cotton F., Desmurs-Clavel H. et al. Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006;253(10):1267-77. DOI: 10.1007/s00415-006-0160-9.
24. Nicolazzi M.A., Carnicelli A., Fuorlo M. et al. Cardiovascular involvement in Erdheim-Chester disease: a case report and review of the literature. Medicine (Baltimore) 2015t;94(43):e1365. DOI: 10.1097/MD.0000000000001365.
25. Lee K., Kim H.R., Roh J. et al. Erdheim- Chester disease presenting as an anterior mediastinal tumor without skeletal involvement. Korean J Thorac Cardiovasc Surg 2018;51(3):223-6. DOI: 10.5090/kjtcs.2018.51.3.223.
26. Razanamahery J., Jacquier F., Humbert S. A rare case of chylous ascites. Gastroenterology 2017;153(4):903-5. DOI: 10.1053/j.gastro.2017.04.008.
27. Balasubramanian G., Modiri A., Affi M. et al.A fatal case of Erdheim-Chester disease with hepatic involvement. ACG Case Rep J 2017;4:e95. DOI: 10.14309/crj.2017.95.
28. Ambrosini V., Savelli F., Merli E. et al. F-18 FDG PET/CT detects muscle involvement in Erdheim-Chester disease. Clin Nucl Med 2012;37(2):196-7. DOI: 10.1097/RLU.0b013e31823e9d54.
29. Martineau P., Pelletier-Galarneau M., Zeng W. The imaging findings of Erdheim-Chester disease: a multimodality approach to diagnosis and staging. World J Nucl Med 2017;16(1):71-4. DOI: 10.4103/1450-1147.181149.
30. Sabino D., do Vale R.H.B., Duarte P.S. et al. Complementary findings on 18F-FDG PET/CT and 18F-NaF PET/CT in a patient with Erdheim-Chester disease. Radiol Bras 2017;50(3):202-3. DOI: 10.1590/0100-3984.2015.0172.
33. Boissel N., Wechsler B., Leblond V. Treatment of refractory Erdheim-Chester disease with double autologous hematopoietic stem-cell transplantation. Ann Intern Med 2001;135(9):844-5.
34. Gaspar N., Boudou P., Haroche J. et al. Highdose chemotherapy followed by autologous hematopoietic stem cell transplantation for adult histiocytic disorders with central nervous system involvement. Haematologica 2006;91(8):1121-5.
35. Braiteh F., Boxrud C., Esmaeli B., Kurzrock R. Successful treatment of Erdheim-Chester disease, a non-Langerhans-cell histiocytosis, with interferonalpha. Blood 2005;106(9):2992-4. DOI: 10.1182/blood-2005-06-2238.
36. Haroche J., Amoura Z., Trad S.G. et al. Variability in the efficacy of interferon-alpha in Erdheim-Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 2006;54(10):3330-6. DOI: 10.1002/art.22165.
37. Arnaud L., Hervier B., Néel A. et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 2011;117(10):2778-82. DOI: 10.1182/blood-2010-06-294108.
38. Aouba A., Georgin-Lavialle S., Pagnoux C. et al. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood 2010;116(20):4070-6. DOI: 10.1182/blood-2010-04-279240.
39. Aubert O., Aouba A., Deshayes S. et al. Favorable radiological outcome of skeletal Erdheim-Chester disease involvement with anakinra. Joint Bone Spine 2013;80(2):206-7. DOI: 10.1016/j.jbspin.2012.07.005.
40. Goyal G., Shah M.V., Call T.G. et al. Efficacy of biological agents in the treatment of Erdheim-Chester disease. Br J Haematol 2018;183(3):520-4. DOI: 10.1111/bjh.14997.
41. Tomelleri A., Cavalli G., De Luca G. et al. Treating heart inflammation with interleukin-1 blockade in a case of Erdheim-Chester disease. Front Immunol 2018;9:1233. DOI: 10.3389/fimmu.2018.01233
43. Nikonova A., Esfahani K., Chausse G. et al. Erdheim-Chester disease: the importance of information integration. Case Rep Oncol 2017;10(2):613-9. DOI: 10.1159/000477658.
44. Hyman D.M., Puzanov I., Subbiah V. et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373(8):726-36. DOI: 10.1056/NEJMoa1502309.
45. Mirouse A., Savey L., Domont F. et al. Systemic vasculitis associated with vemurafenib treatment: case report and literature review. Medicine (Baltimore) 2016;95(46):e4988. DOI: 10.1097/MD.0000000000004988.
46. Bhatia A., Ulaner G., Rampal R. et al. Single-agent dabrafenib for BRAFV600E-mutated histiocytosis. Haematologica 2018;103(4):e177-80. DOI: 10.3324/haematol.2017.185298.
47. Cohen Aubart F., Emile J.F., Carrat F. et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood 2017;130(11):1377-80. DOI: 10.1182/blood-2017-03-771873.
50. Dagna L., Corti A., Langheim S. et al. Tumor necrosis factor α as a master regulator of inflammation in Erdheim-Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol 2012;30(28):e286-90. DOI: 10.1200/JCO.2012.41.9911.
51. Cives M., Simone V., Rizzo F.M. et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015;95(1):1-11. DOI: 10.1016/j.critrevonc.2015.02.004.
53. Azadeh N., Tazelaar H.D., Gotway M.B. et al. Erdheim-Chester disease treated successfully with cladribine. Respir Med Case Rep 2016;18:37-40. DOI: 10.1016/j.rmcr.2016.03.008.
54. Бялик Т.Е., Якимович О.Ю., Махонова Л.А. и др. Диссеминированная ювенильная ксантогранулема у взрослых. Клиническое наблюдение. Клиническая онкогематология 2011;4(4):329-33.
55. Poiroux L., Paycha F., Polivka M., Ea H.K. Efficacy of zoledronic acid in Erdheim-Chester disease: a case report. Joint Bone Spine 2016;83(5):573-5. DOI: 10.1016/j.jbspin.2015.10.010
Болезнь Бехтерева
Анкилозирующий спондилит (спондилоартрит) — хроническое воспалительное заболевание межпозвоночных суставов, которое заканчивается полной неподвижностью позвоночника. Его называют в народе болезнью Бехтерева — по фамилии врача-психоневролога, который впервые определил признаки недуга. Ученые связывают патологию с генетическими и наследственными причинами, а также — особенностями иммунной системы реагировать на внешние раздражители.
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обратиться к профильному специалисту.

Симптомы
Начинаться спондилоартрит может с шейного или поясничного отдела резко либо исподволь. В первом случае боль «выстреливает» из шеи в руку или из поясницы в ногу. Во втором — проявления напоминают остеохондроз или радикулит. Вначале пациент может жаловаться на болезненные ощущения в пояснице. Они обычно на время проходят после массажа, упражнений, прогревания. Без лечения дискомфорт постепенно нарастает. Его характер становится следующим:
- усиление болей после трех часов ночи;
- утихание после полудня;
- утренняя скованность движений в пояснице.
У пятидесяти процентов больных наблюдаются повышение температуры тела, покраснение и сухость белков глаз, похудение. С течением болезни позвоночник и грудная клетка быстро теряют подвижность. Дыхание становится поверхностным, в легких появляются застойные явления, что провоцирует воспаления в дыхательных путях. Движения делаются все более затрудненными, внешний вид больного характеризуется следующими признаками:
- отсутствие естественного изгиба позвоночника;
- сильная сутулость;
- ноги слегка согнуты в коленях
Больной не может нагнуться, повернуться вправо или влево. Его позвоночник напоминает монолит, полностью лишенный гибкости. Чтобы осуществить какие-либо действия, он должен повернуться всем телом.
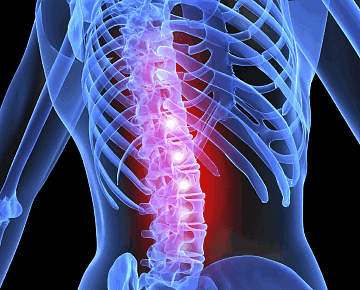
Причины
Анкилозирующий спондилит считается аутоиммунным заболеванием. При нем нарушаются защитные механизмы организма, который воспринимает собственные клетки как чужие и начинает борьбу против них, вырабатывая антитела. Мотивы появления спондилоартрита до конца еще не выяснены. Предполагается, что на развитие заболевания влияют наследственная предрасположенность либо генетическая особенность, причем, вызвать ее может именно ген HLA-B27. Но даже те, кто подвержен болезни, могут избежать ее при отсутствии следующих факторов:
- инфекционные заболевания вирусной природы;
- частые переохлаждения;
- травмы опорно-двигательного аппарата;
- простуды;
- скрытые мочеполовые или кишечные инфекции.
В последнее время ученые доказали, что очень часто недуг возникает на фоне психосоматических явлений, таких как:
- затяжные стрессы;
- продолжительные нервные нагрузки;
- повышенное чувство ответственности;
- подавление негативных эмоций;
- хронические депрессии.
В развитии болезни играет роль наличие некроза опухоли ФНО-а — специфического белка, активного в отношении иммунных процессов. Такой белок может провоцировать воспаления крестцово-подвздошного соединения.
Диагностика
Спондилоартрит необходимо отличать от патологий позвоночника дегенеративного характера, что будет влиять на лечебную тактику. С этой целью врач обязательно направляет больного на следующие методы обследования:
- Рентгенография позвоночника и крестца. Способ очень эффективный, снимок покажет воспаления и первые признаки окостенения позвонков. Его точность приближается к 95 %. Стоимость рентгена позвоночника в московских клиниках начинается от 1800 рублей. Цена на снимок крестца — от 417 рублей.
- Анализ крови на воспалительный процесс. Внимание следует обратить на скорость оседания эритроцитов. При воспалении показатель подскочит до 35 миллиметров в час. Цена на общий анализ крови в клиниках Москвы колеблется от 295 до 1168 рублей.
- Молекулярно-генетическое исследование крови на выявление антигена HLA-B27. Материал берется из вены. Непосредственно перед сдачей анализа не рекомендуется курить. Средняя стоимость услуги в медицинских центрах столицы составляет примерно 3024 рубля.
К какому врачу обратиться?
Если вы подозреваете у себя болезнь Бехтерева, важно не допустить ее прогрессирования, поэтому лечение нужно начать вовремя. При «прострелах» в конечности либо болях в пояснице следует обратиться к специалисту, которым является:
Читайте также: