Синдром Жанбона (Janbon) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 21.01.2026
Московский научно-практический центр дерматологии и косметологии Департамента здравоохранения Москвы, Москва, Россия, 119071
ГКБ №14 им. В.Г. Короленко Департамента здравоохранения Москвы
Семейный случай синдрома Джанотти—Крости
Журнал: Клиническая дерматология и венерология. 2015;14(4): 26‑29
Представлено редкое клиническое наблюдение синдрома Джанотти—Крости. Его необычность заключается в семейном характере — одновременном заболевании у матери и двух ее детей. Синдром отличает клинический полиморфизм, что затрудняет его диагностику.
Первые описания синдрома Джанотти—Крости (СД-К — акродерматит папулезный детей) относятся к 1955 и 1956 гг. Его характеризует возникновение мономорфных эритематозных папул у детей в акральных зонах: на конечностях, лице и шее [1]. Появление этих элементов связывали с гепатитом, обусловленным HBsAg [2]. Как оказалось, возбудителями синдрома могут быть также вирусы Коксаки А-16, инфекционного мононуклеоза, кори, краснухи, гепатитов, А и С, Эпштейна—Барр, парагриппа, ротавирус, аденовирус, энтеровирусы, парвовирусы, цитомегаловирус, бактериальные инфекты (Mycoplasma pneumoniaе, Borrelia burgdorferi, Bartonella hensеlаe, b-гемолитический стрептококк). В эту группу входят и вакцины против гриппа, дифтерии, столбняка, коклюша [3—9]. Иногда заболевание возникает после перенесенного ОРВИ, тонзиллита, фарингита, бронхита, энтероколита [10].
В настоящее время СД-К рассматривают как ответную реакцию организма на различные инфекционные агенты [4, 7, 11, 12]. Хотя считают, что дерматоз редко встречается в популяции, допускается, что заболевание имеет широкое распространение, но из-за неосведомленности врачей не диагностируется [5, 6, 11].
Болеют преимущественно дети, чаще мальчики в возрасте от 3 мес до 15 лет. Пик частоты возникновения СД-К приходится на возраст 1—6 лет [4—6, 11]. Встречаются единичные случаи заболевания взрослых [6]. Обычно болезнь развивается спорадически в течение года без определенной сезонности [2]. Редко синдром проявляется в эпидемической форме [2, 4].
Развитию экзантемы может предшествовать продром в виде респираторных симптомов, фарингита, субфебрилитета [6, 7, 9]. Обычно болезнь возникает остро, в течение нескольких дней, проявляясь мономорфными полусферическими или плоскими папулами на стопах, голенях, бедрах, ягодицах. Затем процесс распространяется на предплечья, кисти, лицо [7, 9—11]. У младенцев папулы имеют диаметр 5—10 мм розового или насыщенного красного цвета, располагаются сгруппированно; у детей постарше диаметр эффлоресценций составляет 2—4 мм [2, 4]. Сыпь нередко располагается симметрично. Характерно слияние элементов сыпи (папул, везикул) в бляшки, а также образование пурпуры, инфильтрации [2, 5, 11].
Иногда высыпаниям сопутствует легкое недомогание, диспепсия, диарея, гепатоспленомегалия, лимфаденит (паховый, аксилярный), в крови повышается уровень трансаминаз, лейкопения, моноцитоз, гипохромная анемия [3, 5, 9, 11]. Субъективно процесс мало беспокоит [3, 8, 12]. Патогистологические изменения не специфичны [10].
Дифференциальную диагностику СД-К проводят с красным плоским лишаем, каплевидным парапсориазом, полиморфной экссудативной эритемой, розовым лишаем, токсико-аллергическим дерматитом, герпетиформной экземой Капоши, папулезной крапивницей, ксантоматозом, инфекционным мононуклеозом, мелкоузелковым саркоидозом [5, 8, 10].
Прогноз заболевания благоприятный [4]. Так как синдром имеет тенденцию к спонтанному разрешению через 2—8 нед [4, 9—11], в большинстве случаев лечение не требуется. Эффективные меры профилактики отсутствуют [2, 8].
Зачастую диагноз основывается на клинической оценке анамнеза, характера высыпаний и их течения.
В консультативное отделение МНПЦДК ДЗМ филиала «Центр детской дерматологии и косметологии» обратилась мать с двумя детьми: мальчиком А., 4 лет, и девочкой М., 2 лет, с жалобами на высыпания на коже щек, живота, верхних и нижних конечностей, сопровождающихся незначительным зудом, преимущественно в вечернее время. Начало заболевания ни с чем не связывали. Первые высыпания появились около 1 нед назад у мальчика на коже щек и живота. Через 2 дня процесс распространился на кожу верхних и нижних конечностей. На следующий день появились аналогичные высыпания у матери и девочки. За медицинской помощью не обращались. Несколько дней лечились самостоятельно кремом бороплюс (без положительного эффекта).
Status localis: кожный процесс носил распространенный, симметричный островоспалительный характер; локализовался на коже щек, разгибательной поверхности верхних и нижних конечностей; был представлен розоватыми, отечными, полусферическими папулами, размером от 1 до 5 мм, плотноватыми при пальпации. Эффлоресценции располагались на фоне эритемы, отечности; очаги имели четкие границы (рис. 1).
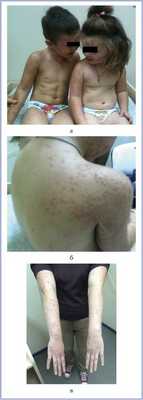
Рис. 1. Синдром Джанотти—Крости. Высыпания у брата и сестры (а, б) и их матери (в).
Клинический анализ крови: у мальчика относительный лимфоцитоз (71%), повышенная СОЭ (13 мм/час); у девочки эозинофилия (16%).
Аналогичные, но менее выраженные высыпания наблюдались у матери (см. рис. 1, в). Они локализовались на коже лба, разгибательной поверхности верхних и нижних конечностей. Были представлены диссеминированными, не склонными к слиянию, четко очерченными папулами, диаметром 4 мм, ярко-розового цвета. Несколько уплотненные узелки незначительно возвышались над кожей.
На основании клинических проявлений, характера элементов сыпи, течения заболевания диагностирован СД-К.
На фоне проводимой терапии (супрастин, аскорутин, кальция глюконат перорально, лосьон каламин, эмолиум, эмульсия для тела, суспензия циндол наружно) процесс регрессировал в течение 3 нед. В большинстве очагов высыпания полностью разрешились. На коже разгибательной поверхности плеч после их регресса остались бледно-розовые пятна (рис. 2). Только у матери на коже лица, верхних и нижних конечностей сохранялись папулезные элементы.
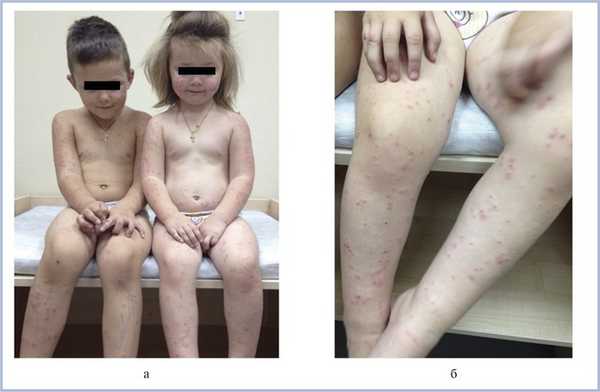
Рис. 2. Эти же больные в динамике.
Представленное нами клиническое наблюдение семейной формы СД-К (одновременное заболевание троих членов одной семьи) представляет для дерматологов несомненный интерес. Своевременная диагностика, адекватное (симптоматическое) лечение способствуют скорому разрешению патологического процесса.
Баршоня — Тешендорфа синдром (Th. Barsony, 1887-1942, венгерский рентгенолог; W. Teschendorf, немецкий рентгенолог, XX в.; синонимы: дивертикулы пищевода множественные ложные, дивертикулы пищевода множественные функциональные, пищевод извитой, пищевод четкообразный, пищевод штопорообразный) — болезнь пищевода неизвестной этиологии, характеризующаяся спастическим сокращением его циркулярных мышц, что придает пищеводу четкообразный вид на рентгенограмме; клинически проявляется непостоянной дисфагией и загрудинными болями.
Бойса симптом (Boys) — появление булькающего звука или урчания при надавливании на боковую поверхность шеи; признак большого дивертикула шейного отдела пищевода.
Бушара болезнь (Ch.J. Bouchard, 1837-1915, французский патолог) — гастрэктазия (расширение полости желудка с растяжением его стенок при стенозе привратниковой части или двенадцатиперстной кишки).
Бэррета язва пищевода (N.R. Barret, род. в 1903 г., английский хирург) — язва пищевода, по клинико-морфологическим признакам напоминающая пептическую язву желудка или двенадцатиперстной кишки.
Кушинга эзофагит (Н.W. Cushing, 1869-1939, американский нейрохирург) — острый эзофагит, иногда развивающийся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кушинга язва (Н.W. Cushing) — язва желудка или двенадцатиперстной кишки, иногда развивающаяся при поражении ЦНС, напр. после черепно-мозговой травмы.
Кэмерона синдром (A.J. Cameron) — язвенные или эрозивные повреждения слизистой оболочки грыжевого выпячивания при грыжах пищеводного отверстия диафрагмы, которые сопровождаются хроническим оккультным или явным кровотечением и железодефицитной анемией.
Ларрея грыжа (D.J. Larrey, 1766-1842, французский хирург). Левосторонняя парастернальная диафрагмальная грыжа, выходящая в средостение через грудино-реберный треугольник.
Лихтенштерна симптом (Lichtenstern, синоним дисфагия парадоксальная, d. paradoxalis) — дисфагия, при которой большие порции пищи легче проходят в желудок, чем малые; возможный признак грыжи пищеводного отверстия диафрагмы.
Лиана — Сигье — Вельти синдром (С.С. Lian, 1882-1969, французский врач; F. Siguier, 1909-1972, французский врач; Н.L. Welti, 1895-1970, французский хирург) — сочетание диафрагмальной грыжи, часто осложненной рефлюкс-эзофагитом, с повторными тромбозами и тромбофлебитами сосудов конечностей и нередко с гипохромной анемией; патогенез неясен.
Монтандона синдром (А. Montandon, швейцарский врач, XX век) — приступы длительной дисфагии вследствие миогенного стеноза шейной части пищевода (области пищеводноглоточного соединения), сопровождающиеся попаданием жидкой пищи в гортань, аспирации и приводящие к нарастающему истощению.
Морганьи грыжа (G.B. Morgagni, 1682-1771, итальянский анатом). Правосторонняя парастернальная грыжа.
Харрингтона грыжа (S. W. Harrington, 1889-1975, американский хирург). Антральная грыжа пищеводного отверстия диафрагмы (типы 1 и 2).
Ценкера дивертикул (F.A. Zenker, 1825-1898, немецкий патолог; синоним ценкеровский дивертикул) — мешковидный дивертикул глоточного конца пищевода, образующийся сначала на его задней стенке, а затем распространяющийся и на боковую. В начале заболевания — глоточные парестезии, перемежающаяся дисфагия, сухой кашель. С увеличением дивертикула в нем происходит задержка пищи, урчание при приеме воды. Мешок сдавливает пищевод, усиливается дисфагия, часты аспирационные пневмонии.
Эпонимные симптомы/синдромы, связанные с патологией кишечника
Алапи симптом (Alapy). Отсутствие или незначительное напряжение брюшной стенки при инвагинации кишечника.
Альвареса синдром (W.C. Alvarez, 1884-1978, американский врач, описан в 1949 г.; синонимы: псевдоилеус; истерическое вздутие живота). Преходящее вздутие живота нейрогенной природы. Чаще наблюдается у истеричных или психопатичных женщин.
Бувре признак (1) (L. Bouveret, 1850-1929, французский врач) — выпячивание в области проекции на переднюю брюшную стенку места перехода подвздошной кишки в слепую, наблюдаемое при непроходимости толстой кишки.
Бувре признак (2) (L. Bouveret; синоним Куссмауля симптом, Adolf Kußmaul, 1822-1902, немецкий терапевт) — периодическое выбухание брюшной стенки в надчревной области и левом подреберье при сужении привратника, обусловленное усиленной перистальтикой желудка.
Вербрайка синдром (J.R. Verbryke, род. в 1885 г., американский врач; синоним синдром печеночного перегиба ободочной кишки) — боль и чувство натяжения в подложечной области в сочетании с диспептическими явлениями при наличии сращений желчного пузыря с печеночным углом ободочной кишки.
Вильмса симптом падающей капли (М. Wilms, 1867-1918, немецкий хирург и онколог) — звук падающей капли жидкости, определяющийся аускультативно на фоне шумов перистальтики при непроходимости кишечника.
Гарднера синдром (Е.J. Gardner, 1909-1989, американский врач-генетик) — наследственная болезнь, характеризующаяся множественным полипозом прямой и ободочной кишок в сочетании с доброкачественными опухолями, чаще костей и кожи (остеомы, фибромы, липомы); наследуется по аутосомно-доминантному типу.
Гейбнер-гертеровская болезнь (O.J.L. Heubner, 1843-1926, немецкий педиатр; С.A. Herter, 1865-1910, американский врач и фармаколог; синоним Ги — Гертера — Гейбнера болезнь) — целиакия.
Данса симптом (J. В. H. Dance, 1797-1832, французский врач) — западение брюшной стенки в правой подвздошной области при илеоцекальной инвагинации.
Жанбона синдром (М. Janbon, современный французский терапевт; синоним syndromus choleriformis, enteritis choleriformis). Описан как гастроинтестинальная симптоматика после лечения окситетрациклином. Патогенез заключается в уничтожении антибиотиками физиологической бактериальной флоры кишечника, что способствует развитию патогенной, устойчивой к антибиотикам.
Ирасека — Цюльцера — Уилсона синдром (A. Jirasek, 1880-1960, чехословацкий хирург; W.W. Zuelzer, 1909-1987, американский педиатр; J.L. Wilson, американский педиатр) — аганглиоз толстой кишки врожденный сегментарный. Патоморфологическая форма болезни Гиршспрунга, при которой в толстой кишке имеются одна или две аганглионарные зоны с нормальным участком кишки между ними; при этом аганглиоз не распространяется на прямую кишку.
Кантора симптом (М.О. Cantor, род. в 1907 г., американский хирург) — нитевидные тени в дефектах наполнения кишки; рентгенологический признак колита и регионарного илеита.
Карно симптом (M.O. Carnot) — боль в эпигастральной области, возникающая при резком разгибании туловища. Бывает при спаечной болезни.
Кенига синдром (F. Konig, 1832-1910, немецкий хирург) — сочетание приступов коликообразных болей в животе, метеоризма, чередования запоров и поносов, урчания при пальпации правой подвздошной ямки, наблюдающееся при хронической непроходимости кишечника в области перехода подвздошной кишки в слепую.
Клойбера симптом (Н. Kloiber, немецкий рентгенолог; синоним Клойбера чаши) — наличие на рентгенограмме живота (при вертикальном положении больного) теней, напоминающих чаши с жидкостью; признак скопления жидкости и газа в кишечнике при его непроходимости.
Кронкхайта — Канада синдром (L.М. Cronkhite, род. в 1919 г., американский врач; W.J. Canada, современный американский рентгенолог) — облысение, атрофия ногтей и гиперпигментация кожи при семейном полипозе органов пищеварительного тракта.
Кюсса синдром (G.Е. Kuss, 1877-1967, французский хирург) — хроническая частичная непроходимость кишечника, обусловленная наличием спаек в области малого таза, например при хроническом сальпингите.
Лобри — Сулля синдром (Ch. Laubry, 1872-1941, французский врач; P.L.J. Soulle, 1903-1960, франц. врач) — сочетание избыточного содержания газа в желудке, метеоризма и высокого стояния правого купола диафрагмы при ишемической болезни сердца, обусловленное рефлекторной гипокинезией пищеварительного тракта.
Мачеллы — Дворкена — Биля симптом (T.E. Machella, F.J. Dworken, H.J. Biel; 1952). Синоним: симптом селезеночного угла. Сильная боль в левой половине живота, вызванная растяжением газами селезеночного угла толстой кишки. Облегчение наступает после опорожнения кишечника и отхождения газов. См. Пайра болезнь, синдром.
Матье симптом (A. Mathieu, 1855-1917, французский врач) — шум плеска в пупочной области при толчкообразной пальпации; признак непроходимости кишечника.
Меккеля дивертикул (J.F. Meckel junior, 1781-1833, немецкий анатом). Врожденный дивертикул подвздошной кишки. Правило двоек: 2 дюйма длиной, в 2 футах от илеоцекального клапана (баугиниевой заслонки), у 2% населения.
Образцова признак (В.П. Образцов, 1849-1920, отечественный терапевт) — шум плеска при пальпации слепой кишки; признак хронического колита.
Обуховской больницы симптом — баллонообразное расширение ампулы прямой кишки, определяемое пальцевым исследованием; признак заворота сигмовидной кишки.
Пайра болезнь, синдром (Erwin Payr, австрогерманский хирург, 1871-1946; синонимы: синдром селезеночного изгиба, flexura lienalis syndrome, splenic flexure syndrome). Первично описана как запоры, обусловленные перегибом и сращением поперечной и нисходящей ободочной кишок («двустволка»). Разнообразная симптоматика (в т. ч. кардиалгии) связана с скоплением газа в области изгиба. Одни считают вариантом СРК (облегчение после опорожнения кишечника и отхождения газов), другие - самостоятельной патологией. Иногда называют также синдромом Мачеллы-Дворкена-Била.
Пиулахса — Хедериха (Пиулакса - Эдерика) болезнь, синдром (испанские врачи P. Piulachs, H. Hederich; синоним: тимпанит при синдроме долихомегаколона, tympanites in dolichomegacolon syndrome). Острое паралитическое расширение толстой кишки (без механической непроходимости). Симптомы: резкий метеоризм и боль в животе. Рентгенологически обнаруживают долихомегаколон.
Протопопова синдром (В.П. Протопопов, 1880-1957, отечественный психиатр; синоним Протопопова триада) — сочетание тахикардии, мидриаза (расширения зрачка) и спастического запора, наблюдающееся при депрессиях.
Рапунцель синдром (описан E.D. Vaughan, J.L. Sawyers и H.W. Scott в 1968 г.). Закупорка кишок, вызванная систематическим проглатыванием волос. В кишках больного образуются трихобезоары. Может потребовать хирургического вмешательства. Наблюдается при психопатиях, шизофрении, эпилепсии, олигофрении, преимущественно в детском возрасте. Рапунцель (Rapunzel) - персонаж одноименной сказки братьев Гримм, девушка с длинными косами.
Сейнта синдром (Ch.F.М. Saint, совр. южноафриканский патолог; синонимы: Сейнта триада, Сена синдром — нрк*, Сента синдром — нрк*) — сочетание грыжи пищеводного отверстия диафрагмы, желчнокаменной болезни и дивертикулеза толстой кишки.
Тавеля болезнь (Е. Tavevl, швейцарский хирург) — периколит после аппендэктомии, характеризующийся образованием спаек и сужением толстой кишки, лихорадкой и расстройствами функции кишечника.
Miserere! (помилосердствуй!) от начала католической молитвы "miserere mei" - помилуй меня (Боже), так по-старому называлась каловая рвота (copremesis) при непроходимости кишечника.
Синдром Дабина-Джонсона ( Генетически обусловленный пигментный гепатоз , Энзимопатическая желтуха )
Синдром Дабина-Джонсона — это хроническое наследственное заболевание, которое характеризуется нарушением выделения билирубина из гепатоцитов в желчь. Основное клиническое проявление — интермиттирующая желтуха. Для болезни также характерны диспепсические расстройства, снижение аппетита и ухудшение общего самочувствия. Диагностика синдрома Дабина-Джонсона включает биохимические анализы крови и мочи, бромсульфалеиновую пробу, инструментальные методики (УЗИ, лапароскопию, биопсию печени). Лечение предполагает коррекцию образа жизни, назначение щадящей диеты. При необходимости используют желчегонные препараты и другие лекарственные средства.
МКБ-10
Общие сведения
Синдром имеет несколько синонимов — энзимопатическая желтуха, генетически обусловленный пигментный гепатоз. Нозологическая форма названа в честь двух американских ученых — И.Н. Дабина и Ф.Б. Джонсона, описавших синдром в 1954 году. Это редкое заболевание, в 70% проявляющееся в молодом возрасте. В основном синдром Дабина-Джонсона наблюдается у жителей Среднего Востока. Наибольшая частота встречаемости среди иранских евреев — 1 случай на 1300 населения. У 60% больных патология сопровождается снижением активности факторов коагуляции и кровотечениями. Заболеваемость не зависит от пола.
Причины
Синдром имеет генетическую природу, характеризуется аутосомно-рецессивным путем наследования. Среди родственников болезнь может проявляться как у мужчин, так и у женщин, повторяется через 1-2 поколения. Наследственный дефект представлен мутацией в нуклеотидной последовательности, обеспечивающей кодирование белка MRP2. Этот протеин отвечает за экскрецию (выделение) конъюгированного билирубина и органических анионов в желчные ходы.
Патогенез
Вследствие дисфункции АТФ-зависимой транспортной системы канальцев желчь не проходит в желчные капилляры, из-за чего билирубин накапливается в печеночных тканях. Длительно существующий синдром сопровождается обратными биохимическими реакциями: избыточное количество прямого билирубина освобождается от глюкуроновой кислоты. Полученный непрямой билирубин также проникает в кровоток, обуславливает токсическое действие на нервную систему.
Нарушается выделение активных метаболитов эпинефрина (триптофана, тирозина). Как следствие, в печени накапливаются меланиноподобные пигменты, расположенные в лизосомах гепатоцитов. При макроскопическом исследовании в печеночной паренхиме видны множественные темные пятна — так называемая «шоколадная печень». При исследовании образцов под микроскопом наблюдаются скопления пигментных зерен большей частью в центре долек.
Симптомы
Обычно манифестация синдрома Дабина-Джонсона происходит в 20-30-летнем возрасте, хотя первые характерные признаки появляются уже у подростков. Крайне редко заболевание выявляется у детей. У женщин с бессимптомным течением энзимопатической желтухи клинический дебют болезни могут спровоцировать наступление беременности или прием контрацептивных средств.
Основный симптом заболевания — желтуха, не сопровождаемая кожным зудом. Сначала возникает желтушность склер и слизистых оболочек, затем кожа также приобретает желтушный оттенок. Желтуху могут усиливать изнурительные физнагрузки, стрессовые ситуации, интеркуррентные инфекции. Как правило, желтушные периоды сменяются безжелтушными, хотя у ряда пациентов иктеричность кожи сохраняется постоянно.
Синдром периодически обостряется. Пациент жалуется на сильные боли справа в подреберье, реже — в околопупочной области. Иногда боль отдает в правое плечо или лопатку. Болевой синдром может быть настолько интенсивным, что напоминает печеночную колику. Одновременно с болью беспокоят тошнота, горечь во рту. Изредка бывает рвота, которая не приносит облегчения. Общая интоксикация проявляется повышенной утомляемостью, сонливостью, снижением аппетита.
Осложнения
Синдром Дабина-Джонсона отличается доброкачественным течением и при правильном лечении не вызывает неприятных последствий. Частое осложнение при длительно протекающем заболевании — воспалительные процессы в желчном пузыре и протоках. Воспаление может переходить и на печеночную паренхиму, вызывая холестатический гепатит. При наличии этих патологий желтуха у больных синдромом Дабина-Джонсона становится постоянной.
При присоединении бактериальной инфекции может возникать эмпиема желчного пузыря и гнойный холангит, которые при неблагоприятных условиях трансформируются в ограниченный перитонит. У пожилых людей наследственный пигментный синдром провоцирует появление фиброзных изменений в печеночных дольках. В результате снижается функциональная активность печени. Вследствие нарушения синтеза факторов свертывания крови возрастает риск кровотечений.
Диагностика
При физикальном обследовании пациентов врач-гастроэнтеролог или гепатолог пальпирует выступающий на несколько сантиметров из-под реберной дуги край печени. Синдром дифференцируют с болезнью Ротора, гепатитами, раковыми метастазами в печень. Симптомы поражения желчного пузыря (Мюсси, Ортнера, Кера) отрицательные. Для подтверждения заболевания используют следующие лабораторные и инструментальные исследования:
- Анализы крови. Основным диагностическим критерием синдрома является повышение уровня общего билирубина более 85 мкмоль/л, при этом массовая доля прямого билирубина превышает 15%. При выполнении коагулограммы у 60% пациентов выявляют снижение активности протромбина и уменьшение протромбинового времени.
- Анализ мочи. В моче обнаруживают повышенный уровень билирубина. Для дифференциальной диагностики с близким по клинической симптоматике синдромом Ротора оценивают соотношение копропорфиринов I и III типа в моче. При гепатозе Дабина-Джонсона количество копропорфирина 1 типа составляет 80%, а 3 типа — 20%.
- Бромсульфалеиновая проба. Анализ наиболее чувствителен для оценки печеночных функций. При синдроме Дабина-Джонсона спустя 45 минут концентрация в крови специального красителя, который предварительно ввели внутривенно, составляет более 6%. Такая проба считается положительной и указывает на снижение поглотительно-выделительной функции печени.
- УЗИ органов брюшной полости. При сонографии синдром проявляется увеличением печени на 1-2 см, у пациентов среднего и пожилого возраста нередко обнаруживают очаги фиброза. Размеры и контуры желчного пузыря не изменены, конкременты не визуализируются. Характерно одновременное увеличение селезенки.
- Инвазивные методы. В сомнительных ситуациях показана диагностическая лапароскопия, которая позволяет врачу осмотреть наружную поверхность печени и выявить характерные коричневые пятна. Чтобы подтвердить диагноз, назначается чрескожная биопсия печеночной ткани с последующим осмотром биоптатов под электронным микроскопом.
Лечение синдрома Дабина-Джонсона
Этиопатогенетическое лечение заболевания не разработано. Главная роль в предупреждении ухудшений состояния больных принадлежит немедикаментозным мероприятиям. Чтобы предотвратить обострения синдрома Дабина-Джонсона, пациентам рекомендуют по возможности избегать физического переутомления и стрессов. Основные направления лечения, применяемые в современной гастроэнтерологии для улучшения качества жизни человека:
- Диета. В рационе питания ограничивают потребление тугоплавких жиров и продуктов, содержащих консерванты. Полностью исключают употребление спиртных напитков. Показана витаминизированная диета с достаточной калорийностью.
- Препараты с желчегонным эффектом. Назначаются синтетические или растительные лекарственные средства, которые либо увеличивают концентрацию желчных кислот, либо повышают содержание водного компонента желчи. Специалисты отдают предпочтение мягким растительным препаратам.
- Санация очагов инфекции. Проводится выявление и лечение самых распространенных хронических источников инфекции — кариеса зубов и хронического тонзиллита. При обнаружении сопутствующей патологии желчевыводящих путей подбирается соответствующая патогенетическая терапия.
Прогноз и профилактика
Наличие болезни Дабина-Джонсона не влияет на продолжительность жизни и работоспособность, поэтому прогноз благоприятный. Синдром не прогрессирует. При соблюдении врачебных рекомендаций пациенты чувствуют себя хорошо. Больным необходимо воздерживаться от алкоголя и приема контрацептивов. Специфическая профилактика не разработана. Семьям с синдромом Дабина-Джонсона перед планированием беременности необходимо проконсультироваться с врачом-генетиком.
2. Клинические особенности течение синдрома Дабина-Джонсона у детей/ В.С. Березенко, М.Б. Диба, Ю.П. Резников// Современная педиатрия. — 2018.
4. Пигментные гепатозы: клинические особенности, пункционная биопсия, электронная микроскопия, диагноз, прогноз/ С.Д. Подымова// Практическая гастроэнтерология. — 2018.
Синдром Гийена — Барре - симптомы и лечение
Что такое синдром Гийена — Барре? Причины возникновения, диагностику и методы лечения разберем в статье доктора Жуйкова Александра Вячеславовича, невролога со стажем в 21 год.
Над статьей доктора Жуйкова Александра Вячеславовича работали литературный редактор Елена Бережная , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Гийена — Барре (ГБС) — острое аутоиммунное заболевание, которое охватывает группу острых нарушений периферической нервной системы. Характеризуется мышечной слабостью, а также болью и ползанием мурашек в начале болезни из-за поражения чувствительных волокон. Каждый вариант нарушений характеризуется особенностями патофизиологии и клинического распределения слабости в конечностях и черепных нервах.
Распространённость синдрома Гийена — Барре
Синдром Гийена — Барре встречается в 1-2 случаях на 100 000 населения в год. [10]
Причины синдрома Гийена — Барре
Точная причина синдрома Гийена — Барре неизвестна. Но у 70% пациентов с ГБС наблюдались предшествующие инфекционные заболевания: респираторные, желудочно-кишечные инфекции, вирус Зика. Также синдром Гийена — Барре может развиться после заражения коронавирусом. [9]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гийена — Барре
Симптомы ОРВИ или расстройства желудочно-кишечного тракта отмечаются у 2/3 пациентов. Первыми симптомами ГБС являются парестезии пальцев конечностей, за которыми следует прогрессирующая слабость мышц нижних конечностей и нарушения походки. Болезнь прогрессирует в течение нескольких часов или дней, возникает слабость верхних конечностей и развиваются паралич черепных нервов. Параличи обычно симметричны и носят, конечно, периферический характер. У половины пациентов боль может быть первоначальной жалобой, что затрудняет диагноз. Атаксия и боль чаще встречаются у детей, чем у взрослых. Задержка мочи наблюдается у 10%-15% больных. Поражение вегетативных нервов проявляются головокружениями, гипертонией, чрезмерным потоотделением и тахикардией.
При объективном обследовании выявляется восходящая мышечная слабость, а также арефлексия. Сухожильные рефлексы нижних конечностей отсутствуют, но рефлексы верхней конечности могут вызываться. Мышечная слабость может задействовать и респираторные мышцы. Поражение черепных нервов отмечается в 35-50%, вегетативная нестабильность в 26%-50%, атаксия — в 23%, дизестезия — в 20% случаев. [1]
Наиболее распространенными признаками вегетативной дисфункции являются синусовая тахикардия или брадикардия и артериальная гипертония. У пациентов с тяжелой вегетативной дисфункцией наблюдаются изменения периферического вазомоторного тонуса с гипотензией и лабильностью артериального давления.
Нечастые варианты клинического течения болезни включают лихорадку в начале неврологических симптомов, тяжелую сенсорную недостаточность с болью (миалгии и артралгии, менингизм, корешковая боль), дисфункции сфинктеров.
Возможность ГБС должна рассматриваться у любого пациента с быстрым развитием острой нервно-мышечной слабости. На ранней стадии ГБС следует отличать от других заболеваний с прогрессирующей симметричной мышечной слабостью, включая поперечный миелит и миелопатию, острую токсическую или дифтеритическую полиневропатию, порфирию, миастению и нарушения электролитного обмена (например, гипокалиемия).
Патогенез синдрома Гийена — Барре
Нейрофизиологические процессы, лежащие в основе ГБС, подразделяются на несколько подтипов. Наиболее распространенные подтипы включают:
- острую воспалительную демиелинизирующую полирадикулопатию;
- острую двигательную аксональную невропатию;
- острую моторную и сенсорную аксональную нейропатию;
- синдром Миллера-Фишера, как вариант ГБС, характеризуется триадой признаков: офтальмоплегия, атаксия и арефлексия.
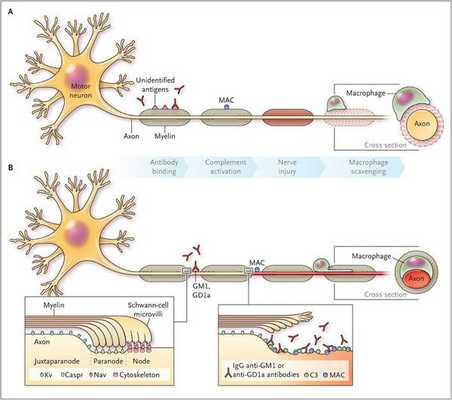
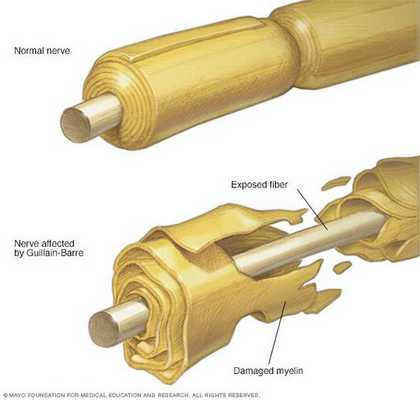
Считается, что ГБС развивается вследствие выработки антител против белка инфекционного агента, которые перекрестно реагируют с ганглиозидами нервных волокон человека. Аутоантитела связываются с миелиновыми антигенами и активируют комплемент, с формированием мембранно-атакующего комплекса на внешней поверхности клеток Шванна. Повреждение оболочек нервных стволов приводит к нарушениям проводимости и мышечной слабости (на поздней стадии может происходить и аксональная дегенерация). Демиелинизирующее поражение наблюдается по всей длине периферического нерва, включая нервные корешки.
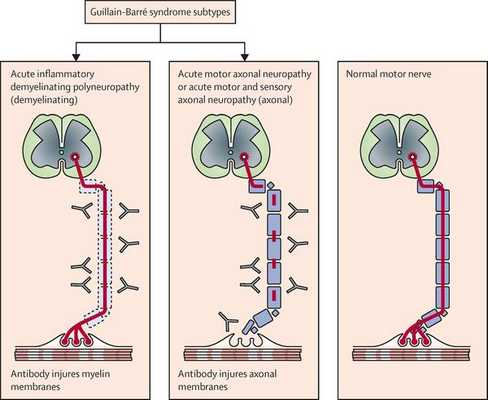
Поражаются все типы нервов, в том числе вегетативные, моторные и сенсорные волокна. Вовлечение двигательных нервов происходит значительно чаще, чем сенсорных.
Классификация и стадии развития синдрома Гийена — Барре
В Международной классификации болезней (МКБ-10) синдром Гийена — Барре кодируется как G61.0.
Основные виды синдрома Гийена — Барре:
- Острая воспалительная демиелинизирующая полирадикулоневропатия (ОВДП) — самая распространённая форма в Северной Америке и Европе. Основным признаком ОВДП является мышечная слабость, которая сперва возникает в нижней части тела, а затем распространяется вверх.
- Синдром Миллера Фишера — проявляется параличом глаз и неустойчивостью походки. Эта форма более распространена в Азии.
- Острая моторная аксональная невропатия (ОМАН) и острая моторно-сенсорная аксональная невропатия (ОМСАН) — чаще встречаются в Китае, Японии и Мексике. [8]
Осложнения синдрома Гийена — Барре
Пациенты с ГБС подвержены риску опасных для жизни респираторных осложнений и вегетативных нарушений.
Показания для перевода в отделение интенсивной терапии включают:
- быстрое прогрессирование моторной слабости с поражением респираторных мышц;
- вентиляционную дыхательную недостаточность;
- пневмонию;
- бульбарные расстройства;
- тяжелую вегетативную недостаточность.
Осложнения проводимого лечения, требующие интенсивной терапии, включают перегрузку жидкостью, анафилаксию на введение внутривенного иммуноглобулина или гемодинамические нарушения при проведении плазмафереза.
У 15%-25% детей с ГБС развивается декомпенсированная дыхательная недостаточность, которая требует механической вентиляции легких. [2] Респираторные нарушения чаще встречается у детей с быстрым прогрессированием заболевания, слабостью верхних конечностей, вегетативной дисфункцией и поражениями черепных нервов. Интубация трахеи может потребоваться у больных для защиты дыхательных путей, проведения механической вентиляции легких. При ГБС быстрое прогрессирование, двусторонний паралич лицевого нерва и вегетативная дисфункция предопределяют повышенную вероятность интубации. Необходимо планирование ранней интубации для минимизации риска осложнений и необходимости проведения экстренной интубации.
Вегетативная дисфункция повышает риск эндотрахеальной интубации. С другой стороны, дисавтономия может увеличить риск гемодинамических реакций на препараты, используемые для индукции анестезии во время интубации.
Признаки, указывающие на необходимость механической вентиляции легких: [4]
- вентиляционная дыхательная недостаточность;
- увеличение потребности в кислороде для поддержания SpO2 выше 92%;
- признаки альвеолярной гиповентиляции (PCO2 выше 50 мм. рт. ст.);
- быстрое снижение жизненной емкости на 50% по сравнению с исходным уровнем;
- невозможность кашля
Вегетативная дисфункция является основным фактором смертности при ГБС. Фатальный сердечно-сосудистый коллапс из-за вегетативной дисфункции наблюдается у 2%-10% тяжелобольных пациентов. [3] Мониторинг частоты сердечных сокращений, артериального давления и электрокардиограммы следует продолжать до тех пор, пока пациенты нуждаются в респираторной поддержке. Чрескожная кардиостимуляция может потребоваться при выраженной брадикардии. Гипотония корректируется восполнением объема циркулирующей крови (ОЦК), и, если пациент невосприимчив к восполнению ОЦК, применяются α-агонисты, такие как норадреналин, мезатон, адреналин.
При нестабильной гемодинамике непрерывная регистрация артериального и центрального венозного давления должна проводиться для контроля объема инфузионной терапии.
Артериальная гипертензия может возникать, но это осложнение не требует специального лечения, если оно не осложняется отеком легких, энцефалопатией или субарахноидальным кровоизлиянием.
Диагностика синдрома Гийена — Барре
Сбор жалоб и анамнеза
На приёме врач в первую очередь обратит внимание на скорость распространения паралича и нарушение дыхания. Если эти признаки выражены, больному может потребоваться экстренная помощь.
Как правило, пациенты с синдромом Гийена — Барре жалуются на нарушение походки, онемение и зябкость ног, а затем и рук. Нередко пациенты рассказывают, что недавно перенесли ОРВИ.
Осмотр
Объективный неврологический осмотр — это основа диагностики при синдроме Гийена — Барре. Врач оценивает рефлексы, координацию движений, походку, чувствительность и мышечную силу.
Лабораторная диагностика
Основным видом лабораторной диагностики при синдроме Гийена — Барре является исследование спинномозговой жидкости, которую получают с помощью люмбальной пункции.
Инструментальная диагностика
ЭНМГ (Электронейромиография) — единственный инструментальный метод диагностики, позволяющий подтвердить диагноз ГБС и уточнить характер патологических изменений (демиелинизирующий или аксональный) и их распространенность. [3]
Игольчатая электромиография характеризуется наличием признаков текущего денервационно-реиннервационного процесса при полинейропатии. Исследуют дистальные мышцы верхних и нижних конечностей (например, переднюю большеберцовую мышцу, общий разгибатель пальцев), а при необходимости и проксимальные мышцы (например, четырёхглавую мышцу бедра).
ЭНМГ-исследование у больных с ГБС зависит от клинических проявлений:
- при дистальных парезах исследуются длинные нервы на руках и ногах: не менее четырех двигательных и четырех чувствительных (двигательные и чувствительные порции срединного и локтевого нервов; малоберцовый, большеберцовый, поверхностный малоберцовый и икроножный нервы с одной стороны).
Оценка основных ЭНМГ- параметров:
Первые признаки денервационного процесса появляются через две-три недели после начала заболевания, признаки реиннервационного процесса — через месяц.
Дифференциальная диагностика
Синдром Гийена — Барре следует отличать от следующих заболеваний:
- и клещевого энцефалита (чувствительность не нарушена, поражены преимущественно черепные нервы); (отягощённый эпидемиологический анамнез, например посещение эндемичных стран); (чувствительность не нарушена, рефлексы снижены незначительно);
- обменно-метаболических полиневропатий (течение хроническое).
Лечение синдрома Гийена — Барре
Показания для госпитализации
Практически во всех случаях требуется госпитализация. Экстренная госпитализация необходима пациентам с нарушениями дыхания — в таких случаях лечение проводят в условиях реанимации.
Общие принципы лечения синдрома Гийена — Барре
Лечение острой демиелинизирующей полирадикулоневропатии комплексное. Основа — плазмаферез, иммуноглобулины и кортикостероиды. В ряде случаев требуется искусственная вентиляция лёгких, коррекция нарушений кровообращения, профилактика инфекционных и тромбоэмболических осложнений. Обязательным условием успешного лечения является уход.
Общее поддерживающее лечение и уход
Пациенты, требующие интенсивной терапии, требуют тщательного общего ухода. Запор наблюдается более чем в 50% случаев пациентов с ГБС в результате динамической непроходимости кишечника. Может потребоваться искусственное питание.
Медикаментозное лечение и плазмаферез
В лечении ГБС предпринимаются различные виды иммуномодулирующей терапии. [1] [2]
Внутривенный иммуноглобулин назначают в виде ежедневной инфузии (в дозе 0,4 гр/кг/день) в течение 5 дней в первые 2 недели болезни. Второй курс иммуноглобулина может потребоваться 5%-10% пациентов, при отрицательной динамике после первоначального улучшения. Механизм действия внутривенного иммуноглобулина, вероятно, многофакторный и, как полагают, включает модуляцию активации комплемента, нейтрализацию идиотипических антител, подавление воспалительных медиаторов (цитокины, хемокины).
Побочные эффекты иммуноглобулина включают головную боль, миалгию и артралгию, гриппоподобные симптомы, лихорадку. У пациентов с дефицитом IgA может развиться анафилаксия после первого курса внутривенного иммуноглобулина.
Плазмаферез способствует удалению антител, вовлеченных в патогенез ГБС. В течение каждого сеанса 40-50 мл/кг плазмы заменяют смесью 0,9% раствора хлорида натрия и альбумина. Проведение плазмафереза приводит к сокращению времени выздоровления и снижению потребности в искусственной вентиляции. Эти преимущества очевидны, если плазмаферез проводится в течение первых двух недель после начала болезни. Осложнения, связанные с плазмаферезом, включают гематому в области венопункции, пневмоторакс после катетеризации подключичной вены и сепсис. Плазмаферез противопоказан пациентам с тяжелой гемодинамической нестабильностью, кровотечением и сепсисом. Комбинация плазмафереза и иммуноглобулина не показала клинических преимуществ.
Симптоматическое лечение синдрома Гийена — Барре:
- при боли применяют парацетамол;
- катадолон и трамадол применяют при выраженном болевом синдроме;
- при нейропатической боли эффективны карбамазепин и габапентин.
Оперативное лечение
При тяжёлом течении может потребоваться длительная респираторная поддержка и наложение трахеостомы. Если пациент находится на искусственном питании, то накладывают гастростому.
Прогноз. Профилактика
ГБС остается серьезным заболеванием, несмотря на улучшение результатов лечения. По сравнению со взрослыми, у детей чаще отмечается более благоприятное течение заболевания, с полным, а не частичным выздоровлением. Причинами неблагоприятного исхода при ГБС являются дыхательная недостаточность, осложнения искусственной вентиляции легких (пневмония, сепсис, острый респираторный дистресс-синдром и тромбоэмболические осложнения), остановка сердца, вторичная по отношению к дисавтономии.
Восстановление обычно начинается через две-четыре недели после прекращения прогрессирования симптомов. Среднее время от начала заболевания до полного выздоровления составляет 60 дней. Данные относительно долгосрочного исхода ГБС ограничены. 75% - 80% пациентов полностью выздоравливают. Около 20% пациентов не могут ходить через полгода.
Младшая возрастная группа (менее 9 лет), быстрое прогрессирование и максимальная мышечная слабость, потребность в искусственной вентиляции легких являются важными предикторами длительного двигательного дефицита. [4]
Клиника с Нуля
Главной целью каждой нашей статьи является информирование о той или иной болезни, о состоянии или синдроме. Мы не пытаемся напугать или запугать, но хотим проинформировать. Предупреждён - значит вооружён. И сегодня мы снова будем вооружать вас полезной и очень важной информацией. Синдром Джанотти-Крости или детский папулёзный дерматит: что это такое, как проявляется и что с ним делать - подробно в новой статье.
Что за синдром?
Синдром Джанотти-Крости - это детский папулёзный акродерматит, который представляет собой сыпь в виде папул, как правило, симметричную, локализующуюся на щеках, ягодицах и сгибах предплечий и ног.
Этот синдром был описан итальянскими дерматологами в 1953-1956 годах Джанотти Ф. и Крости А. Изначально предполагалось, что этот синдром свойственен только младенцам и детям, но позже выяснилось, что у взрослых он тоже может быть.
Изначально авторы, описавшие этот синдром, говорили о трёх кардинальных проявлениях синдрома:
- нерецидивный эритематозно-папулёзный дерматит, который проявляется на лице и конечностях, длится около 3 недель;
- увеличение лимфоузлов;
- острый гепатит, как правило, без желтухи, которые может длится несколько месяцев и прогрессирует в хронические заболевания печени.
Но в нашем времени увеличение лимфоузлов и гепатит не являются обязательными для диагностики синдрома Джанотти-Крости.
Неизвестна частота распространения заболевания. Наравне с этим можно сказать, что синдром недостаточно диагностирован, так как у многих детей может быть «вирусная сыпь» или «неспецифическая вирусная экзантема».
Что характерно для синдрома Джанотти-Крости на сегодняшний день?
- Преимущественно болеют дети от 2 месяцев до 15 лет, при это немногим чаще заболевание диагностируют у мужского пола.
- Взрослые женщины могут быть более склонны к развитию синдрома, нежели мужчины.
- Пик частоты заболевания приходится на детей в возрасте от 6 месяцев до 6 лет. Чем младше возраст, тем больше сыпи.
- Сезонности болезнь не имеет, хотя и было отмечено, что большинство проявлений приходилось на осенне-зимний период.
Каков прогноз? Как правило, благоприятный. Синдром Джанотти-Крости проходит самостоятельно через 2-8 недель. При этом, отмечаются случаи, когда сыпь держалась от 10 дней до 6 месяцев, также сообщалось о продолжительности от 5 дней до 12 месяцев, а зуд может длиться неделями. В большинстве случаев лечения этот синдром не требует.
При появлении первых симптомов, то есть сыпи, первым делом стоит обратиться к педиатру или терапевту, а если будет необходимость, то к дерматологу и инфекционисту.
Синдром Джанотти-Крости возникает после перенесённых вирусных заболеваний, как ответ на неё. Это значит, что возможно за 7-14 дней до первых симптомов ребёнок мог переболеть ОРВИ. Наиболее распространёнными причинами являются вирус гепатита В и вирус Эпштейна-Барр. Стоит отметить, что в тех странах, где проводится всеобщая вакцинация от гепатита В в младенчестве, этот вирус является причиной синдрома Джанотти-Крости крайне редко.
У младенцев и детей младшего возраста с острой инфекцией вируса гепатита В синдром Джанотти-Крости может быть единственным клиническим проявлением, что спустя десятилетия может развиться в хронический гепатит.
На долю вируса Эпштейна-Барр приходится от половины до трёх четвертей всех случаев синдрома.
Также синдром Джанотти-Крости хоть и реже, но возникает на фоне таких микроорганизмов, как энтеровирусы, цитомегаловирус, парвовирус, вирус парагриппа, вирус гепатита А, ротавирус, контагиозный моллюск, реапираторно-синцитиальный вирус, вирус иммунодефицита человека, вирус герпеса человека 6 типа (в основном 6В), микоплазма pneumoniae, бета-гемолитические стрептококки, Bartonella henselae и Borrelia burgdorferi.
Синдром Джанотти-Крости является чрезвычайно редким состоянием после вакцинации, которое проходит самостоятельно. Но не стоит паниковать и отказываться от вакцин полностью, так как несмотря на то, что вакцины не защищают со 100% вероятностью, но совершенно точно защищают от осложнений, которые могут дать всевозможные заболевания, в том числе и от таких, которые угрожают жизни.
Как проявляется?
Синдром Джанотти-Крости характеризуется в первую очередь появлением множественных симметричных папулёзных или папуловезикулярных высыпаний. Для этих высыпаний характерны следующие симптомы:
- мономорфные, то есть, неизменные розовато-коричневые папулы или папуловезикулы с плоским верхом, диаметр которых составляет от 1 до 10 мм;
- могут сливаться в бляшки;
- поражают лицо, ягодицы, разгибательные части предплечий и ног, и ступни;
- при этом, если сыпь есть на туловище, то не является исключением синдрома Джанотти-Крости;
- не поражает слизистые оболочки и ногти;
- на ранней стадии заболевания можно увидеть сыпь в местах травмы;
- сыпь сопровождается зудом от лёгкой степени до средней степени тяжести;
- возможны геморрагические изменения, особенно в областях, подверженных травме;
- возможно недомогание, диарея или повышенная температура в переделах от 37,1 до 38 С;
- возможно увеличение лимфатических узлов, характерно для 25-35% пациентов (как правило, вовлечены шейные, подмышечные и паховые лимфоузлы);
- очень редко встречается патологическое увеличение селезёнки;
- не известно, как часто при синдроме Джанотти-Крости поражается печень, так как при наличии гепатита не появляется желтухи.
Течение заболевания появляется в следующем:
- новые высыпания появляются в первые 2-3 недели, расширяются зоны поражения;
- в средней фазе заболевания распределение высыпаний является наиболее классическим;
- к концу болезни высыпания медленно разрешаются;
- возможно наличие легкой поствоспалительной гиперпигментации или гипопигментации;
- при этом рубцы и изменения пигмента, как правило, проходят и не остаются навсегда;
- синдром может рецидивировать, но редко;
- увеличение лимфоузлов, печени, селезёнки проходят чуть дольше, чем кожная сыпь.
Синдром Джанотти-Крости не заразен и никак не связан с аллергией!
Диагностика синдрома
Для синдрома Джанотти-Крости нет каких-то характерных анализов или тестов. Врач должен обратить внимание на наличие желтухи или на увеличенную печень, чтобы сделать акцент на поиск вируса гепатита В и на поиск повышенного уровня ферментов печени. Но здесь тоже стоит отметить, что наличие повышенных ферментов печени, скорее всего будут говорить о вирусе Эпштейна-Барра.
Некоторые случаи синдрома Джанотти-Крости могут требовать прохождения биопсии кожи.
К слову, вопрос о том, нужны ли младенцам тесты на наличие вируса гепатита В, остаётся спорным. Во-первых, гепатит В, всё-таки редкая причина синдрома Джанотти-Крости в странах, где распространена повсеместная вакцинация от гепатита В в младенческом возрасте. Также в таких развивающихся странах, как Индия тоже можно сказать о том, что гепатит В является редкой причиной синдрома.
Во-вторых, наличие желтухи также не говорит о том, что есть гепатит В, так как при синдроме Джанотти-Крости гепатит В способен протекать без неё.
Но, с другой стороны, синдром Джанотти-Крости может быть единственным проявлением гепатита В у младенцев и детей младшего возраста.
Конечно, необходимо учитывать все факторы при диагностике заболевания, начиная от этнического происхождения, вирус гепатита В в анамнезе у родителей, если это известно, физическое состояние ребёнка, статус иммунизации малыша, а также распространена ли инфекция вируса гепатита В в местности, где живёт ребёнок - все эти факторы важно знать при постановке диагноза.
Осложнения при синдроме Джанотти-Крости встречаются крайне редко.
Лечение синдрома Джанотти-Крости
Специфического лечения синдрома Джанотти-Крости не существует. Как правило, оно состоит в облегчении зуда.
У большинства детей зуд сокращается с применением эмолентов. Врач может назначить местно лосьон каламин для детей, у которых зуд средней степени тяжести.
Если у малыша сильный зуд, то может быть назначен антигистаминный препарат с седативным эффектом.
Возможно применение глюкокортикостероидов при синдроме Джанотти-Крости, хотя они и не могут влиять на течение болезни, но их можно использовать при зуде.
Конечно, лечение назначается только врачом! Только врач сможет провести верную диагностику, провести обследование и расчитать верную дозировку и длительность применения средств.
Подводя итог, хочется сказать о том, что иногда единственное, в чём следует убедить родителей - это тенденция синдрома к самостоятельному разрешению. То есть не важно, что будет использовано для лечения синдрома Джанотти-Крости, на длительность это не повляет. В большинстве случаев всё, что необходимо - это ждать. И самое главное: несмотря на то, что синдром способен проходить самостоятельно, заниматься самолечением не стоит и при появлении симптомов лучше сразу показаться врачу для постановки точного диагноза, так как можно упустить и более тяжёлое заболевание. Берегите себя и своих детей и будьте здоровы!
Читайте также: