Сосудистые пурпуры - виды, классификация
Добавил пользователь Алексей Ф. Обновлено: 01.02.2026
Название патологического процесса, для которого характерно увеличение количества кровеносных или лимфатических сосудов и их изменение, пришло к нам их греческого языка («αγγείο» - сосуд, «δυσπλασία» - неправильное формирование тканей).
Ангиодисплазия может представлять собой врожденную аномалию и диагностироваться у детей, только появившихся на свет, либо дебютировать в любом возрасте (чаще преклонном) в силу различных причин, которые, как правило, остаются невыясненными (по большей части это касается болезней сосудов кишечника).
У детей данная патология нередко бросается в глаза окружающим, поскольку для своей локализации в большинстве случаев выбирает область лица или нижних конечностей, на руках и других частях тела подобные «родимые пятна» (так называют в народе врожденную сосудистую дисплазию, определяемую визуально) отмечаются реже.
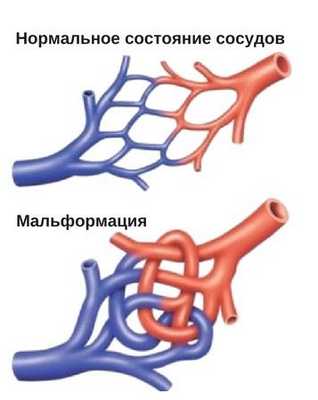
Ангиодисплазия, мальформация или гемангиома?
По МКБ-10 болезнь относят к категории врожденных пороков, однако сосудистую дисплазию кишечника, выявленную у взрослых людей, специалисты определяют как приобретенную форму.
Следует заметить, что до сих пор ученые, занятые исследованием ангиодисплазии, не пришли к единому мнению в отношении применения терминологии, описывающей дефекты сосудов, признаки данной патологии и воспринимают, и трактуют по-разному, поэтому в литературе можно встретить другие названия болезни - врожденная артериовенозная мальформация или даже гемангиома.
Ангиодисплазия является результатом пороков развития кровеносных либо лимфатических сосудов на разных этапах эмбриогенеза, то есть, болезнь берет свое начало во время пребывания и формирования нового организма в утробе матери. Локализация патологического процесса может быть самой разнообразной: видимые части тела, легкие, головной мозг, конечности, желудочно-кишечный тракт и т. д.
Причиной дисплазии сосудов чаще выступают неприятности, постигшие женщину в столь ответственный период (вынашивание) или факторы риска, которые она спровоцировала сама:
Аномалии, формирующиеся на генетическом и хромосомном уровне (обычно это пороки, возникающие вследствие таких факторов риска, как возраст женщины, вредные химические вещества, полученные будущей матерью при разных обстоятельствах в первом триместре беременности);
Все это особенно опасно в период активной закладки кровеносной и лимфатической системы. Правда, в народе бытует мнение, что розовые (красные, синие, фиолетовые, коричневые) пятна, с которыми родился малыш, возникают от того, что женщина во время беременности сильно испугалась и в страхе схватилась за какую-то часть своего тела (“упаси бог”, тронуть руками лицо и тогда ангиодисплазия лица у ребенка обеспечена?). Конечно, это не более, чем « бабские забобоны» (суеверия).
Сложная классификация
Сосудистая дисплазия является врожденным дефектом, однако при рождении выявляется далеко не всегда, поскольку патологическому изменению может подвергаться любая из систем (артериальная, венозная, лимфатическая) и уродства могут быть глубоко спрятаны. При этом не исключается присутствие дефектов на уровне разных сосудов (гемолимфатическая мальформация) или доминирование пороков лишь в одной системе (артериальная либо венозная ангиодисплазия). В связи с этим классифицируют сосудистую дисплазию в зависимости от того, какие сосуды в большей степени подверглись патологическому изменению и, исходя из этого, выделяют несколько форм:
виды сосудистых мальформаций
Разделяют сосудистую мальформацию и по месту расположения, например, в литературе и в жизни можно чаще других встретить понятия: ангиодисплазия лица, кишечника, нижних конечностей.
Ангиодисплазия лица, как правило, заметна с рождения, о ней будет идти речь в разделе «капиллярная ангиодисплазия».
Врожденные аномалии сосудов нижних конечностей у детей также проявляют себя практически сразу после появления на свет: образование красных пятен, кровоподтеков, вытягивание конечностей, увеличение их размеров, повышение температуры тела, постоянный плач.
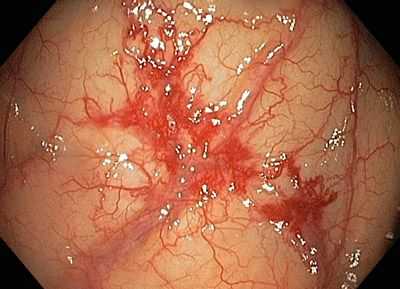
Сложнее обстоят дела с диагностикой сосудистой дисплазии кишечника - при рождении она не видна, кроме этого, многие специалисты склоняются к мысли, что патология носит приобретенный характер, правда, причины в большинстве случаев остаются, как говорится, «за кадром». Между тем, учитывая широкую распространенность патологического изменения сосудов кишечника среди взрослого населения, нельзя обойти вниманием главные симптомы данного процесса, это:
- Кровотечения из заднего прохода, усиливающиеся после физического напряжения;
- Рвота с прожилками крови;
- Примесь крови в кале;
- Боли в животе;
- Признаки анемии (бледность кожи, слабость, сонливость, раздражительность).
Однако, не вдаваясь особо в подробную классификацию, остановимся на основных, наиболее распространенных формах дисплазии сосудов.
Видео: ангиодисплазия толстой кишки
Капиллярная ангиодисплазия
Капиллярная ангиодисплазия (т.н.з. «винные пятна») - наиболее распространенный вид в группе данной патологии (до 1% от всех новорожденных «отмечены» подобным образом).
Капиллярная ангиодисплазия в классическом вариант имеет вид:
- Розоватых пятен, хотя цвет может варьировать от бледно-розового до синюшного и коричневого;
- Пятна не возвышаются над кожными покровами, однако, могут занимать весьма внушительные участки на теле.
Следует отметить, что, хотя и в редких случаях, но капиллярная ангиодисплазия способна создавать немалые проблемы жизнедеятельности всего организма, нарушая отдельные важные функции. Расширяя свои границы по мере роста человека, «родимые пятна» могут препятствовать нормальному функционированию отдельных органов (например, органов зрения - ангиодисплазия лица) либо, располагаясь в местах соприкосновения с одеждой (шея, туловище), могут кровоточить и изъязвляться.
Самое печальное, конечно, если эти «отметины» локализуются на лице, ведь они портят внешность и накладывают отпечаток на характер человека (окружающие люди могут не только останавливать свой взгляд, но еще и комментировать, применяя неприятные выражения).
К счастью, эта врожденная патология сосудов в настоящее время подчиняется лазерному лечению, которое может быть использовано у детей. В результате - девочка или мальчик впоследствии даже могут и не знать об имевшей место аномалии.

Сходная по внешним признакам с капиллярной ангиодисплазией гемангиома, между тем, отлична по своему происхождению. Это - доброкачественная сосудистая опухоль. Она также бывает врожденной, может формироваться в головном мозге и внутренних органах и, если выбирает поверхностное месторасположение, то в большинстве случаев возвышается над кожей (хотя это тоже совсем не обязательно).
Разные формы сосудистой дисплазии
Артериальная ангиодисплазия как изолированный вариант встречается довольно редко, преимущественно она все же наблюдается в комбинации с артериовенозной либо венозной ангиодисплазией. Клинически данная форма проявляется признаками хронической ишемии артериальных сосудов, замедлением роста нижней конечности на участке поражения и нарушением трофики тканей.
Врожденная венозная ангиодисплазия имеет клиническую картину варикозного расширения вен нижних конечностей, хронической венозной недостаточности, нарушения оттока крови и трофических расстройств. В иных случаях пигментные пятна и варикоз могут отсутствовать, однако другие признаки способны поведать о болезни:
- Снижение скорости кровотока, застой крови, увеличение венозного давления;
- Лимфостаз (отеки, увеличение объема конечности, нарушение трофики тканей);
- Повышенная потливость ног (гипергидроз);
- Утолщение рогового слоя эпидермиса и ускоренное его слущивание (гиперкератоз).
К самостоятельному виду венозной ангиодисплазии относят синдром Клиппеля-Треноне с характерным для него поражением исключительно кровеносных сосудов нижних конечностей. Однако классификация данного варианта также противоречива - специалисты опять расходятся во мнениях…
Клиническая картина артериовенозной формы (мальформации) зависит от места расположения сосудистой аномалии. Врожденные, но маленькие по размеру пороки сосудов малого круга кровообращения у детей обычно поначалу остаются незамеченными и «ждут своего часа» до начала прогрессирования. А час их «старта», как правило, совпадает с периодом полового созревания, достигая полного «расцвета» к 20 - 30 годам. И если патологическое изменение касается сосудов малого круга, степень тяжести заболевания находится в зависимости от количества сбрасываемой крови из венозной системы в артериальную. В подобных ситуациях болезнь быстро заявит о себе снижением уровня кислорода в крови - гипоксемией, которая в короткие сроки приобретет хроническую форму. Следует заметить, что клиника артериовенозных мальформаций вообще (в большинстве случаев) тяжелая:
- Повышение температуры в области поражения;
- Дрожание;
- Расширение вен, которое легко обнаружить визуально;
- Явления ишемии и изъязвления, проявляющиеся на кожных покровах;
- Болевые ощущения и периодические кровотечения.
При отсутствии лечения нередко формируется сердечная недостаточность со всеми вытекающими из нее последствиями.
Что касается артериовенозных мальформаций периферических сосудов (нижние конечности, таз, плечевой пояс), то они большей частью заметны при рождении малыша либо в самые ранние годы жизни. Пигментные пятна, варикозное расширение вен, признаки частичного гигантизма не спрячутся от «зоркого ока» родителей и врачей, наблюдающих ребенка.
Лечение
В заключение хочется предупредить читателя, что попытки избавиться от сосудистой дисплазии своими силами и народными средствами ни к чему хорошему не приведут. Лечением всех вариантов данной патологии занимается врач-флеболог и только он может решать, какой метод окажется наиболее оптимальным и действенным. В зависимости от характера патологического процесса пациенту (или родителям) будет предложен один из способов лечения заболевания:

- Лазер - его используют при большой площади поражения с глубоким расположением мальформации;
- Склеротерапия - метод предпочтительный в случае артериовенозных форм;
- Эндоваскулярное лечение - оно часто применяется, если имеют место артериовенозные свищи;
- Эмболизация - метод эффективен при поражении легочных сосудов.
Радикальная хирургия в отношении дисплазии сосудов в настоящее время применяется все реже и реже, ведь после нее остаются рубцы, которые могут выглядеть еще более уродливо, нежели сама болезнь, тем более если её течение бессимптомно.
Геморрагический васкулит ( Аллергическая пурпура , Болезнь Шенлейн-Геноха , Капилляротоксикоз )
Геморрагический васкулит — системное асептическое воспаление сосудов микроциркуляторного русла с преимущественным поражением кожи, суставов, желудочно-кишечного тракта и почечных клубочков. Протекает с явлениями геморрагической или уртикарной сыпи, артралгиями, абдоминальным болевым синдромом, гематурией и почечной недостаточностью. Диагностика основана на клинических симптомах, лабораторных данных (анализ крови, мочи, коагулограмма), исследовании органов ЖКТ и почек. Основой лечения васкулита является терапия антикоагулянтами, ангиагрегантами. В тяжелых случаях применяется экстракорпоральная гемокоррекция, глюкокортикоидная терапия, противовоспалительное, цитостатическое лечение.
МКБ-10

Общие сведения
Геморрагический васкулит (ГВ, болезнь Шенлейн-Геноха, аллергическая пурпура, капилляротоксикоз) относится к наиболее распространенным на сегодняшний день геморрагическим заболеваниям. По сути своей он является аллергическим васкулитом поверхностного характера с поражением мелких артериол, венул, а также капилляров. В Международной классификации болезней (МКБ) заболевание имеет название "аллергическая пурпура". Болезнь Шенлейн-Геноха встречается в основном в детском возрасте - от 5 до 14 лет. Средняя распространенность среди детей этого возраста составляет 23-25 случая на 10 тыс. Наиболее подвержены заболеванию лица в возрасте 7-12 лет. У детей до 3 лет известны лишь отдельные случаи возникновения пурпуры.

Причины
Этиологические аспекты изучены не до конца, известно лишь, что в большинстве случаев патология носит инфекционно-аллергическую природу. Существует сезонная зависимость ‒ наибольшая заболеваемость регистрируется в сырое и холодное время года. Многолетние наблюдения позволили выявить общие триггерные факторы, предшествующие развитию клинических проявлений. К их числу относят:
Во многих наблюдениях причинный фактор, вызвавший возникновение васкулита, установить не удается. Ряд авторов высказывает предположение, что воздействие провоцирующих факторов приводит к развитию геморрагического васкулита лишь в тех случаях, когда оно осуществляется на фоне генетической предрасположенности организма к гиперергическим иммунным реакциям.
Патогенез
В основе механизма развития геморрагического васкулита лежит образование иммунных комплексов и повышение активности белков системы комплемента. Циркулируя в крови, они откладываются на внутренней поверхности стенки мелких сосудов (венул, артериол, капилляров), вызывая ее повреждение с возникновением асептического воспалительного процесса. Воспаление сосудистой стенки в свою очередь приводит к повышению ее проницаемости, отложению в просвете сосуда фибрина и тромботических масс, что обуславливает основные клинические признаки заболевания — кожно-геморрагический синдром и микротромбирование сосудистого русла с поражением ЖКТ, почек, суставов.
Классификация
В клиническом течении капилляротоксикоза различают острую фазу (начальный период или обострение) и фазу стихания (улучшение). По преобладающим симптомам заболевание классифицируют на следующие клинические формы: простую, ревматоидную (суставную), абдоминальную и молниеносную. В соответствии с характером течения различают острый (до 2-х мес.), затяжной (до полугода) и хронический ГВ. По тяжести клинических проявлений выделяют васкулит:
- Легкой степени. Отмечается удовлетворительное состояние пациентов и необильный характер сыпи, артралгии.
- Средней степени. Состояние больного средней тяжести, высыпания обильные, артралгии сопровождаются изменениями в суставах по типу артрита, отмечаются периодические боли в животе и микрогематурия.
- Тяжелой степени. Имеет место тяжелое состояние больного, сливные обильные высыпания с некротическими участками, ангионевротические отеки, нефротический синдром, наблюдается макрогематурия и желудочно-кишечные кровотечения, возможно развитие острой почечной недостаточности.
Симптомы
Для клиники аллергической пурпуры типично острое начало с повышением температуры до субфебрильных или фебрильных цифр. Однако возможно отсутствие подъема температуры. Кожный синдром отмечается в самом дебюте заболевания и наблюдается у всех больных. Он характеризуются диффузными пятнисто-папулезными геморрагическими элементами различной величины (чаще мелкими), не исчезающими при надавливании. В некоторых случаях наблюдается уртикарная сыпь. Высыпания обычно располагаются симметрично на коже голеней, бедер и ягодиц, в области крупных суставов, реже — на коже рук и туловища. Обильность высыпаний часто коррелирует с тяжестью васкулита. При наиболее тяжелом его течении в центре некоторых элементов сыпи развивается некроз и образуется язва. Разрешение сыпи заканчивается длительно сохраняющейся гиперпигментацией. При хроническом течении ГВ с частыми рецидивами на коже после исчезновения сыпи возникает шелушение.
Суставной синдром развивается у 70% пациентов. Поражения суставов могут носить кратковременный характер в виде легкой артралгии или сохраняться в течение нескольких дней с выраженным болевым синдромом, сопровождающимся другими симптомами артрита (покраснение, отечность) и приводящим к ограничению движений в суставе. Типичным является летучий характер поражения с вовлечением преимущественно крупных суставов, чаще коленных и голеностопных. Суставной синдром может появиться в начальном периоде васкулита или возникнуть позже. Зачастую он имеет преходящий характер и никогда не приводит к стойкой деформации суставов. Абдоминальный синдром может предшествовать кожно-суставным проявлениям или сопутствовать им. Он проявляется болями в животе различной интенсивности - от умеренных до приступообразных по типу кишечной колики. Пациенты часто не могут указать точную локализацию боли, жалуются на нарушения стула, тошноту и рвоту. Абдоминалгии могут появляться несколько раз в течение суток и проходят самопроизвольно или в первые несколько дней лечения.
Почечный синдром возникает у 25-30% пациентов и проявляется признаками хронического или острого гломерулонефрита с различной степенью гематурии. У ряда больных возникает нефротический симптомокомплекс. Поражение других органов при геморрагическом васкулите происходит довольно редко. Это может быть геморрагическая пневмония в виде кашля с прожилками крови в мокроте и одышки, кровоизлияния в эндокард, геморрагический перикардит, миокардит. Поражение сосудов головного мозга проявляется головокружением, раздражительностью, головной болью, эпиприступами и может вызвать развитие геморрагического менингита.
Осложнения
Поражение почек является самым стойким синдромом геморрагического васкулита, может осложняться злокачественным гломерулонефритом и хронической почечной недостаточностью. В тяжелых случаях аллергической пурпуры возникают желудочно-кишечные кровотечения, сопровождающиеся кровавой рвотой и присутствием крови в каловых массах, легочные кровотечения, кровоизлияния в вещество головного мозга (геморрагический инсульт). Массивные кровопотери могут привести к коллапсу и анемической коме. Осложнения абдоминального синдрома встречаются реже и представлены инвагинацией кишечника, перитонитом, тромбозом брыжеечных сосудов, некрозом части тонкого кишечника. Наибольшая частота летальных исходов регистрируется при молниеносной форме ГВ.
Диагностика
Проводя диагностику, ревматолог учитывает возраст пациента, изучает этиофакторы, сопоставляет клинические и лабораторные данные, исключает другие заболевания. При развитии почечного синдрома пациенту необходима консультация нефролога, при наличии абдоминальных болей - консультация гастроэнтеролога и хирурга. Диагностическая панель включает:
- Гематологические тесты. В общем анализе крови, как правило, отмечаются неспецифические признаки умеренного воспаления (лейкоцитоз и небольшое повышение СОЭ), увеличение количества тромбоцитов и эозинофилов. Биохимический анализ крови показывает увеличение иммуноглобулина А и СРБ. Большое диагностическое значение имеют результаты коагулограммы. Отсутствие в ней данных за нарушение свертывания при наличии клинических признаков геморрагического синдрома свидетельствует в пользу ГВ.
- Анализы мочи и кала. В анализе мочи выявляется гематурия, протеинурия, цилиндрурия. Пациентам с почечным синдромом показан мониторинг изменений в анализе мочи, проведение биохимии мочи, пробы Зимницкого, Нечипоренко. Для диагностики скрытого ЖКТ-кровотечения производят анализ кала на скрытую кровь.
- Инструментальную диагностику. С целью оценки состояния органов-мишеней выполняется УЗИ почек, УЗДГ почечных сосудов. Для исключения органических причин кровотечения из пищеварительного тракта и бронхов целесообразно проведение УЗИ брюшной полости, гастроскопии, бронхоскопии.
- Биопсию с гистологией. В тяжелых диагностических случаях показана биопсия кожи или почек. Гистологическое исследование биоптата выявляет характерные изменения: отложения иммуноглобулина А и ЦИК на эндотелии и в толще сосудистой стенки венул, артериол и капилляров; образование микротромбов; выход элементов крови за пределы сосуда.
Абдоминальную форму геморрагического васкулита следует дифференцировать от других причин, обуславливающих появление симптомов «острого живота»: аппендицита, пенетрации язвы желудка, острого холецистита, панкреатита, перфорации кишечника при язвенном колите др. Также необходимо исключить тромбоцитопеническую пурпуру, геморрагический синдром при инфекционных заболеваниях (геморрагических лихорадках, гриппе), лейкоз, ревматоидный артрит, болезнь Стилла, острый гломерулонефрит, системные васкулиты.
В острой фазе геморрагического васкулита пациентам необходимо соблюдать постельный режим и гипоаллергенную диету, ограничить употребление жидкости и соли, исключить прием антибиотиков и других медикаментов, которые могут усиливать сенсибилизацию организма. Основные направления терапии зависят от клинических проявлений, поэтому их целесообразно рассматривать посиндромно:
- При любых синдромах. Основу базисной терапии при всех формах ГВ составляет назначение дезагрегантов (дипиридамола, пентоксифиллина) и активаторов фибринолиза (никотиновой кислоты). Препараты этих групп препятствуют агрегации тромбоцитов, улучшают микроциркуляцию и внутритканевую перфузию. Часто в базисную схему включают гепарин и другие антикоагулянты.
- При кожном синдроме. Терапия предполагает применение сульфасалазина, колхицина. Использование преднизолона до сих пор является спорным вопросом среди врачей. Возможно его назначение в тяжелых случаях ГВ. При отсутствии эффекта от терапии кортикостероидами препаратами запаса являются цитостатики.
- При суставном синдроме. Выраженные артралгии купируются проведением противовоспалительной терапии (индометацин, ибупрофен). Дополнительно могут назначаться производные аминохинолина (хлорохин).
- При почечном синдроме. Назначаются высокие дозы глюкокортикоидов, цитостатиков. Возможно использование иАПФ, антагонистов рецепторов ангиотензина II, введение нормального человеческого иммуноглобулина, проведение электрофореза с никотиновой кислотой и гепарином на область почек. В терминальной стадии ХПН требуется гемодиализ или трансплантация почки.
- При абдоминальном синдроме. Интенсивный болевой синдром служит показанием к внутривенному введению преднизолона, реополиглюкина, кристаллоидов. При развитии хирургических осложнений (перфорация, инвагинация кишки) применяется хирургическая тактика.
Тяжелое течение заболевания является показанием для проведения экстракорпоральной гемокоррекции (гемосорбция, иммуносорбция, плазмаферез). Многие авторы отмечают неэффективность антигистаминных препаратов в лечении ГВ. Однако их применение может быть оправдано у пациентов с аллергическим анамнезом. При связи заболевания с пищевой аллергией и наличием абдоминального синдрома дополнительно назначаются энтеросорбенты.
Прогноз и профилктика
Легкие формы геморрагического васкулита склонны к самопроизвольному излечению после первой же атаки заболевания - их прогноз благоприятен. При молниеносной форме смерть пациентов может произойти в первые несколько суток от начала заболевания. Чаще всего это связано с поражением сосудов ЦНС и возникновением внутримозгового кровоизлияния. Другой причиной летального исхода может стать тяжелый почечный синдром, приводящий к развитию уремии. В целях профилактики аллергического васкулита рекомендуется санация хронических инфекционных очагов ЛОР органов, дегельминтизация при глистных инвазиях, исключение контакта с известными аллергенами и бесконтрольного приема медикаментов.
Геморрагические диатезы
Геморрагические диатезы - общее название ряда гематологических синдромов, развивающихся при нарушении того или иного звена гемостаза (тромбоцитарного, сосудистого, плазменного). Общими для всех геморрагических диатезов, независимо от их происхождения, являются синдром повышенной кровоточивости (рецидивирующие, длительные, интенсивные кровотечения, кровоизлияния различных локализаций) и постгеморрагический анемический синдром. Определение клинической формы и причин геморрагических диатезов возможно после всестороннего обследования системы гемостаза - проведения лабораторных тестов и функциональных проб. Лечение включает гемостатическую, гемотрансфузионную терапию, местную остановку кровотечений.


Геморрагические диатезы - болезни крови, характеризующиеся наклонностью организма к возникновению спонтанных или неадекватных травмирующему фактору кровоизлияний и кровотечений. Всего в литературе описано свыше 300 геморрагических диатезов. В основе патологии лежат количественные либо качественные дефекты одного или нескольких факторов свертывания крови. При этом степень кровоточивости может варьировать от мелких петехиальных высыпаний до обширных гематом, массивных наружных и внутренних кровотечений.
По приблизительным данным, в мире около 5 млн. населения страдает первичными геморрагическими диатезами. С учетом вторичных геморрагических состояний (например, ДВС-синдрома), распространенность геморрагических диатезов поистине велика. Проблема осложнений, связанных с геморрагическими диатезами, находится в поле зрения различных медицинских специальностей - гематологии, хирургии, реаниматологии, травматологии, акушерства и гинекологии и мн. др.
Классификация геморрагических диатезов
Геморрагические диатезы принято различать в зависимости от нарушения того или иного фактора гемостаза (тромбоцитарного, коагуляционного или сосудистого). Этот принцип положен в основу широко используемой патогенетической классификации и в соответствии с ним выделяют 3 группы геморрагических диатезов: тромбоцитопатии, коагулопатии и вазопатии.
Тромбоцитопении и тромбоцитопатии, или геморрагические диатезы, связанные с дефектом тромбоцитарного гемостаза (тромбоцитопеническая пурпура, тромбоцитопении при лучевой болезни, лейкозах, геморрагической алейкии; эссенциальная тромбоцитемия, тромбоцитопатии).
Коагулопатии, или геморрагические диатезы, связанные с дефектом коагуляционного гемостаза:
- с нарушением первой фазы свертывания крови - тромбопластинообразования (гемофилия)
- с нарушением второй фазы свертывания крови - превращения протромбина в тромбин (парагемофилия, гипопротромбинемии, болезнь Стюарта Прауэр и др.)
- с нарушением третьей фазы свертывания крови - фибринообразования (фибриногенопатии, врожденная афибриногенемическая пурпура)
- с нарушением фибринолиза (ДВС-синдром)
- с нарушением коагуляции в различных фазах (болезнь Виллебранда и др.)
Вазопатии, или геморрагические диатезы, связанные с дефектом сосудистой стенки (болезнь Рандю-Ослера-Вебера, геморрагический васкулит, авитаминоз С).
Причины геморрагических диатезов
Различают наследственные (первичные) геморрагические диатезы, манифестирующие в детском возрасте, и приобретенные, чаще всего являющиеся вторичными (симптоматическими). Первичные формы являются семейно-наследственными и связаны с врожденным дефектом или дефицитом обычно одного фактора свертывания. Примерами наследственных геморрагических диатезов служат гемофилия, тромбастения Гланцмана, болезнь Рандю-Ослера, болезнь Стюарта Прауэр и др. Исключение составляет болезнь Виллебранда, являющаяся полифакторной коагулопатией, обусловленной нарушением фактора VIII, сосудистого фактора и адгезивности тромбоцитов.
К развитию симптоматических геморрагических диатезов обычно приводит недостаточность сразу нескольких факторов гемостаза. При этом может отмечаться уменьшение их синтеза, повышение расходования, изменение свойств, повреждение эндотелия сосудов и пр. Причинами повышенной кровоточивости могут служить различные заболевания (СКВ, цирроз печени, инфекционный эндокардит), геморрагические лихорадки (лихорадка денге, Марбург, Эбола, Крымская, Омская и др.), дефицит витаминов (С, К и др.). В группу ятрогенных причин входит длительная или неадекватная по дозе терапия антикоагулянтами и тромболитиками.
Чаще всего приобретенные геморрагические диатезы протекают в форме синдрома диссеминированного внутрисосудистого свертывания (тромбогеморрагического синдрома), осложняющего самые различные патологии. Возможно вторичное развитие аутоиммунных, неонатальных, посттрансфузионных тромбоцитопений, геморрагического васкулита, тромбоцитопенической пурпуры, геморрагического синдрома при лучевой болезни, лейкозах и т. д.
Симптомы геморрагических диатезов
В клинике различных форм гемостазиопатий доминируют геморрагический и анемический синдромы. Выраженность их проявлений зависит от патогенетической формы геморрагического диатеза и сопутствующих нарушений. При различных видах геморрагических диатезов могут развиваться разные типы кровотечений.
Микроциркуляторный (капиллярный) тип кровоточивости встречается при тромбоцитопатиях и тромбоцитопениях. Проявляется петехиально-пятнистыми высыпаниями и синяками на коже, кровоизлияниями в слизистые оболочки, кровотечениями после экстракции зуба, десневыми, маточными, носовыми кровотечениями. Геморрагии могут возникать при незначительном травмировании капилляров (при надавливании на кожу, измерении АД и пр.).
Гематомный тип кровоточивости характерен для гемофилии, возможен при передозировке антикоагулянтов. Характеризуется образованием глубоких и болезненных гематом в мягких тканях, гемартрозов, кровоизлияний в подкожно-жировую и забрюшинную клетчатку. Массивные гематомы приводят к расслоению тканей и развитию деструктивных осложнений: контрактур, деформирующих артрозов, патологических переломов. По происхождению такие кровотечения могут быть спонтанными, посттравматическими, послеоперационными.
Капиллярно-гематомные (смешанные) геморрагии сопровождают течение ДВС-синдрома, болезни Виллебранда, наблюдаются при превышении дозы антикоагулянтов. Сочетают петехиально-пятнистые кровоизлияния и гематомы мягких тканей.
Микроангиоматозный тип кровоточивости встречается при геморрагическом ангиоматозе, симптоматических капилляропатиях. При этих геморрагических диатезах возникают упорные рецидивирующие кровотечения одной или двух локализации (обычно носовые, иногда - желудочно-кишечные, легочные, гематурия).
Васкулитно-пурпурный тип кровоточивости отмечается при геморрагических васкулитах. Представляет собой мелкоточечные геморрагии, как правило, имеющие симметричное расположение на конечностях и туловище. После исчезновения кровоизлияний на коже длительно сохраняется остаточная пигментация.
Частые кровотечения вызывают развитие железодефицитной анемии. Для анемического синдрома, сопровождающего течение геморрагических диатезов, характерны слабость, бледность кожных покровов, артериальная гипотония, головокружения, тахикардия. При некоторых геморрагических диатезах может развиваться суставной синдром (припухлость сустава, артралгии), абдоминальный синдром (тошнота, схваткообразные боли), почечный синдром (гематурия, боли в пояснице, дизурия).
Целью диагностики геморрагических диатезов служит определение его формы, причин и степени выраженности патологических сдвигов. План обследования пациента с синдромом повышенной кровоточивости составляется гематологом совместно с лечащим специалистом (ревматологом, хирургом, акушером-гинекологом, травматологом, инфекционистом и др.).
В первую очередь исследуются клинические анализы крови и мочи, количество тромбоцитов, коагулограмма, кал на скрытую кровь. В зависимости от полученных результатов и предполагаемого диагноза назначается расширенная лабораторная и инструментальная диагностика (биохимическое исследование крови, стернальная пункция, трепанобиопсия). При геморрагических диатезах, имеющих иммунный генез, показано определение антиэритроцитарных антител (тест Кумбса), антитромбоцитарных антител, волчаночного антикоагулянта и др. Дополнительные методы могут включать функциональные пробы на ломкость капилляров (пробы жгута, щипка, манжеточную пробу и др.), УЗИ почек, УЗИ печени; рентгенографию суставов и др. Для подтверждения наследственной природы геморрагических диатезов рекомендуется консультация генетика.
Лечение геморрагических диатезов
При подборе лечения практикуется дифференцированный подход, учитывающий патогенетическую форму геморрагического диатеза. Так, при повышенной кровоточивости, вызванной передозировкой антикоагулянтов и тромболитиков, показана отмена данных препаратов или коррекция их дозы; назначение препаратов витамина К (викасола), аминокапроновой кислоты; переливание плазмы. Терапия аутоиммунных геморрагических диатезов основана на применении глюкокортикоидов, иммунодепрессантов, проведении плазмафереза; при нестабильном эффекте от их применения требуется проведение спленэктомии.
При наследственном дефиците того или иного фактора свертываемости показано проведение заместительной терапии их концентратами, трансфузий свежезамороженной плазмы, эритроцитарной массы, гемостатической терапии. С целью местной остановки небольших кровотечений практикуется наложение жгута, давящей повязки, гемостатической губки, льда; проведение тампонады носа и пр. При гемартрозах выполняются лечебные пункции суставов; при гематомах мягких тканей - их дренирование и удаление скопившейся крови.
Основные принципы лечения ДВС-синдрома включают активное устранение причины данного состояния; прекращение внутрисосудистого свертывания, подавление гиперфибринолиза, проведение заместительной гемокомпонентной терапии и т. д.
Осложнения и прогноз
Наиболее частым осложнением геморрагических диатезов служит железодефицитная анемия. При рецидивирующих кровоизлияниях в суставы может развиться их тугоподвижность. Сдавление массивными гематомами нервных стволов чревато возникновением парезов и параличей. Особую опасность представляют профузные внутренние кровотечения, кровоизлияния в головной мозг, надпочечники. Частое повторное переливание препаратов крови является фактором риска развития посттрансфузионных реакций, заражения гепатитом В, ВИЧ-инфекцией.
Течение и исходы геморрагических диатезов различны. При проведении адекватной патогенетической, заместительной и гемостатической терапии прогноз относительно благоприятный. При злокачественных формах с неконтролируемыми кровотечениями и осложнениями исход может быть фатальным.
Тромбоцитопеническая пурпура, идиопатическая: лечение у детей, взрослых
Тромбоцитопеническая пурпура относится к геморрагическим диатезам, которые идут с нарушением тромбоцитарного компонента гемостаза, где количество кровяных пластинок падает ниже допустимого уровня (150 х 10 9 /л). Подобное явление возникает при обстоятельствах, которые способствуют тому, что тромбоциты начинают усиленно разрушаться, чрезмерно потребляться или недостаточно пролиферироваться в костном мозге.
Чаще всего тромбоцитопения (ТП) имеет место при усиленном разрушении клеток, хотя все эти процессы также не исключаются у одного пациента, они сочетаются между собой, идут параллельно и, естественно, усугубляют ситуацию. Количество тромбоцитов в таких случаях падает до критических цифр, что в свою очередь, определяет тяжесть заболевания.
Предпосылки возникновения тромбоцитопении
Большинство тромбоцитопений, как уже доказано, носит приобретенный характер, то есть, генетически запрограммированные дефекты не являются основой заболевания, хотя единичные случаи наследственной патологии все же иной раз имеют место:
- Нарушение синтеза тромбоцитопоэтинов в человеческом организме связывают с наследственной тромбоцитопенией;
- Недостаток ферментов, осуществляющих гликолиз или цикл Кребса, также принадлежит к генетическим аномалиям.
Все остальные состояния, характеризующиеся снижением тромбоцитарного звена, разделяют на иммунные и неиммунные, которые имеют свои определенные причины.
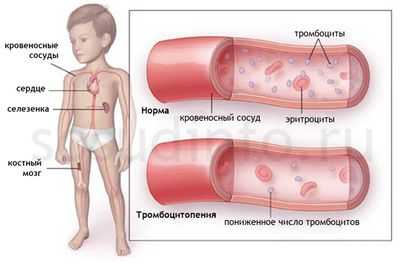
Неиммунные тромбоцитопении
Стартом для возникновения неиммунной тромбоцитопении являются следующие факторы:
- Механическое воздействие на тромбоциты, вызывающее их травмирование (протезирование сосудов, спленомегалии, гемангиомы гигантских размеров);
- Опухоли с метастазами в костный мозг;
- Нарушение кроветворения, сопровождаемое вялым размножением клеток, что характерно для апластической анемии, которой часто сопутствует снижение гемоглобина;
- Воздействие радиации или химических соединений с повреждением миелопоэза;
- Высокая потребность в тромбоцитах при нехватке фолиевой кислоты или Vit В12, диссеминированной агрегации тромбоцитов - ДАТ, РДС - респираторном дистресс-синдроме, тромбозах, ДВС-синдроме, длительной терапии малыми дозами гепарина).
К неиммунному варианту ТП отнесена и тромботическая тромбоцитопеническая пурпура (ТТП), которая имеет острое начало и отличается злокачественным течением. Этиология этого заболевания до сих пор не ясна, однако то, что оно, как правило, ведет к смертельному исходу, известно. Замечено, что ТТП возникает чаще у взрослых в следующих случаях:
- Перенесенная бактериальная или вирусная инфекция;
- Прививки;
- Наличие ВИЧ-инфекции;
- Беременность;
- Применение пероральных противозачаточных средств;
- Лечение некоторыми противоопухолевыми препаратами;
- Коллагенозы;
- Как наследственная патология (очень редко).
Тромботическая тромбоцитопеническая пурпура характеризуется отложением гиалиновых тромбоцитарных тромбов, причиной образования которых стала самопроизвольная агрегация тромбоцитов в сосудах мелкого калибра, в результате чего они закрывают сосуд. Тромбоцитарные тромбы захватывают весь организм человека и повреждают микрососуды многих органов, поэтому ТТП характеризует наличие симптомов:
- Гемолитической анемии;
- Лихорадки;
- Неврологической симптоматики;
- Оострой почечной недостаточности.
Смерть больного обычно наступает в результате отказа почек работать (ОПН).
Выработка антитромбоцитарных антител - путь к иммунной тромбоцитопении
Иммунная тромбоцитопеническая пурпура имеет несколько видов:
- Изоиммунная (аллоиммунная) тромбоцитопения, возникающая часто при внутриутробном развитии. Это происходит, если антитромбоцитарные антитела транспортируются ребенку от матери (снижение тромбоцитов наблюдается у малыша и в первый месяц жизни), или антитела против тромбоцитов появляются в результате переливания крови. А ввиду того, что антитела тромбоцитов относятся к нециркулирующим, они тут же приклеиваются к тромбоцитарным антигенам.
- Гетероиммунная (гаптеновая) тромбоцитопения формируется при образовании антител, которые вырабатываются как ответная реакция на измененную антигенную структуру тромбоцитов (изменяются антигены тромбоцитов и они становятся «чужими» для собственной системы иммунитета), что иногда случается после перенесенных респираторных вирусных заболеваний или после приема некоторых медикаментозных средств. Тромбоцитопения, связанная с вирусами и лекарствами может наблюдаться до полугода, но если за этот промежуток времени она не исчезает, то ее переименовывают и считают уже аутоиммунной.
- Аутоиммунная тромбоцитопения (АИТП), включает наибольшее количество форм, к которым отнесена и так называемая идиопатическая тромбоцитопеническая пурпура, причина которой неизвестна. Идиопатическая форма любого происхождения раньше называлась болезнью Верльгофа, что не применимо к иммунной форме. Болезнью Верльгофа теперь именуют только тромботическую тромбоцитопеническую пурпуру, которая также является идиопатической, но носит неиммунный характер.
Аутоиммунная тромбоцитопения тоже имеет свое деление в зависимости от направленности антител и причины возникновения. Аутоиммунный тромбоцитолиз идиопатическим называют тогда, когда причина агрессии против собственных клеток не установлена, симптоматическим, если удается установить, почему кровяные пластинки вдруг начинают разрушаться. Симптоматическая тромбоцитопеническая пурпура зачастую является спутницей хронических патологических состояний:
Геморрагический диатез при АИТП
- Хронических форм лейкозов (чаще хр. лимфолейкоза);
- Воспалительных заболеваний печени и почек;
При АИТП иммунная система вдруг начинает не узнавать свой родной вполне нормальный во всех отношениях тромбоцит и, принимая его за «чужака», отвечает выработкой на него антител.
Иммунная тромбоцитопеническая пурпура встречается в любом возрасте, начиная с неонатального периода, поэтому у детей она далеко не редкость. Поражает болезнь преимущественно лиц женского пола. Течение патологического процесса часто приобретает хроническую рецидивирующую форму, особенно это касается идиопатической тромбоцитопенической пурпуры, поскольку существующие гипотезы ее возникновения так и не объясняют истинную причину появления болезни.
Развитие пурпуры
Развитие тромбоцитопенической пурпуры во многом зависит от тромбоцитолиза (гибели клеток при влиянии антител). В костном мозге начинают активно продуцироваться мегакариоциты, которые быстро расходуются и увеличивают количество тромбоцитов, поступающих в кровяное русло, где кровяные пластинки гибнут в течение короткого времени. Вместо положенной им недели жизни, они существуют несколько часов, заставляя тем самым костный мозг интенсивно работать и восполнять потери. Такие тромбоцитопении называются гиперрегенераторными, которые преимущественно встречаются у детей и составляют большую часть клинических форм в педиатрии. Но бывает, что антитела, помимо тромбоцитов, направляются и на мегакариоциты, что опустошает росток и не дает возможности кровяным пластинкам образоваться. Это так называемая гипорегенераторная тромбоцитопения, которая не обязательно будет иммунной.
Большая роль в патогенезе ТП отводится и функциональным особенностям тромбоцитов, их участию в гемостазе и питании сосудистой стенки, а также адгезивно-агрегационной способности, ведь они умеют приклеиваться друг к другу и к поврежденному эндотелию с образованием тромбоцитарной пробки.
Можно сделать вывод, что главным моментом, запускающим кровоточивость, является тромбоцитопения. Когда сосудистые стенки перестают получать тромбоцитарную подкормку, наступает их дистрофия, которая не может препятствовать прохождению через сосуды эритроцитов. Малейшая травма в подобных случаях может вызвать длительное кровотечение.
Заподозрить такой диагноз как тромбоцитопеническая пурпура можно, если присутствуют частые носовые кровотечения и геморрагическая петехиально-пятнистая сыпь, которая от аллергической отличается тем, что не исчезает при надавливании. Сниженное количество тромбоцитов в анализе крови подтверждает диагноз ТП.
При диагностике ТП в гемостазиограмме можно получить увеличение времени кровотечения по Дьюку до 30 минут и более и уменьшение (менее 60%) ретракции кровяного сгустка, тогда как свертываемость по Ли Уайту будет оставаться нормальной. Тромбоцитопеническую пурпуру дифференцируют с наследственной тромбоцитопатией (тромбоцитопенией) с помощью семейного анамнеза. Для наследственной тромбоцитопатии характерно сокращение жизни кровяных пластинок за счет неполноценности мембран или недостаток ферментов в самих клетках.
Геморрагическая сыпь - значит, пурпура
Для тромбоцитопенической пурпуры характерно появление петехиально-пятнистого типа кровоточивости. А в случае больших травм могут наблюдаться экхиматозы. Таким образом, симптомы тромбоцитопенической пурпуры можно представить в следующем виде:
- Кровоизлияния, появившиеся в местах инъекций;
- Выраженные кровотечения из слизистых оболочек (ротовая полость, миндалины, глотка);
- Инфекция за одну-две недели до появления сыпи;
- Нормальная температура тела даже у детей и, лишь за редким исключением, она может подняться до субфебрильной;
- Единичные или множественные спонтанные кровоизлияния (иногда после незначительного травмирования);
- Ассиметричное поражение кожи, петехии и «синяки» разных размеров;
- Кровоизлияния разного цвета: от пурпурной (ярко-красная) до сине-зеленой и желтой;
- Синяки рассасываются до 3 недель;
- Несоответствие травмы и кровоизлияния;
- Появление кровоизлияний в ночной период (во время сна);
- Появление геморрагической сыпи на ногах, руках и туловище;
- Кровотечения из носа, десен и в естественные полости;
- Желудочно-кишечные кровотечения (черная окраска стула или примесь алой крови);
- Кровавая рвота, которая носит вторичный характер, так как возникает в результате заглатывания крови из носа;
- Анемия, формирующаяся на почве постоянной потери крови;
- Возможны кровоизлияния в головной мозг, что является весьма опасным симптомом.
Кроме этого в медицинской практике были описаны кровотечения из ушей, кровохарканье и кровоизлияние в стекловидное тело глаза, которое привело к полной слепоте.
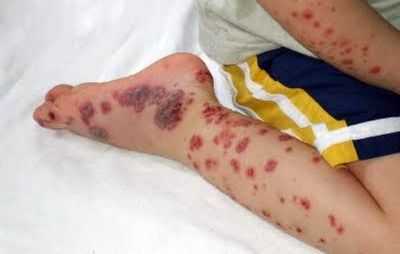
Проявления тромбоцитопенической пурпуры
Тромбоцитопеническую пурпуру люди часто путают с пурпурой Шенлейна-Геноха, для которой характерен васкулитно-пурпурный тип сыпи, поэтому болезнь называют геморрагическим васкулитом. С толку сбивает то, что сыпь похожа на петехиально-пятнистую при ТП. Болезнь Шенлейна-Геноха можно описать таким образом:
- Ярко-красная папулезная сыпь, тускнеющая со временем и оставляющая синеватые пигментные пятна;
- Ощущение зуда перед высыпанием;
- Нередко поднимается температура тела;
- Высыпания располагаются симметрично на ногах и руках;
- Поражение сосудов почек (микро- и макрогематурия).
Сыпь в случае геморрагического васкулита очень напоминает аллергическую, однако при надавливании она не исчезает. Болезнь Шенлейна-Геноха имеет хроническое течение, где, помимо кожи, могут поражаться суставы, желудочно-кишечный тракт, слизистые, поэтому выделяют 4 формы этого заболевания:
- Кожную:
- Абдоминальную;
- Суставную;
- Смешанную.
Как лечить тромбоцитопеническую пурпуру?
Заподозрив тромбоцитопеническую пурпуру, больного следует госпитализировать, ведь строгий постельный режим с подобным заболеванием обязателен до того, пока тромбоциты не восстановятся до минимального физиологического уровня.
Если есть кровотечение, первым делом применяются местные (ε-аминокапроновая кислота, гемостатическая губка, тромбин, адроксон) и общие (аскорутин и кальций хлористый для внутривенного введения) кровоостанавливающие средства. На первом этапе лечебных мероприятий включают кортикостероидную терапию, которая продолжается до 3-х месяцев.
Лечением тромбоцитопенической пурпуры путем переливания тромбомассы увлекаются не очень, ввиду того, что тромбоциты донора должны еще прижиться у реципиента, а не иммунизировать его еще больше (показан индивидуальный подбор), поэтому при глубокой анемии, возникшей на фоне кровопотери, предпочтение отдают отмытым эритроцитам.
Спленэктомия (радикальный метод) производится на втором этапе лечения в случаях упорных кровотечений, асептического воспаления или угрозы разрыва селезенки. Однако если удаление селезенки также не дает эффекта, то лечение продолжают малыми дозами кортикостероидов. Они хоть и не восстановят число тромбоцитов, но, по крайней мере, уменьшат опасность кровоизлияния в головной мозг.
Таким больным абсолютно противопоказаны барбитураты, кофеин, аспирин и другие лекарственные препараты, способствующие снижению тромбоцитарного компонента в крови, поэтому пациент строго предупреждается об этом.
После прохождения курса лечения и выписки из больницы, пациент ставится на диспансерный учет в поликлинике по месту жительства для дальнейшего наблюдения. Обязательной в таком случае является санация всех очагов хронической инфекции, а полости рта - в особенности. Также проводится дегельминтизация.
Учитывая то, что тромбоцитопеническая пурпура не является редкостью у детей, часть ответственности за дальнейшее течение болезни возлагается на родителей. С ними проводится беседа о том, что может спровоцировать рецидив заболевания (ОРВИ, обострение очаговых инфекций). Кроме того, родители должны знать, как постепенно вводить закаливание, лечебную физкультуру и вести пищевой дневник (устранение аллергогенных продуктов). Чтобы уберечь ребенка от травм, он на этот период освобождается от занятий в школе, ему показана учеба в домашних условиях.
На диспансерном учете по выздоровлению человек находится не менее 2-х лет. Прогноз заболевания, если это не тромботическая тромбоцитопеническая пурпура, как правило, благоприятный.
Тромбоцитопеническая пурпура ( Болезнь Верльгофа )
Тромбоцитопеническая пурпура - это разновидность геморрагического диатеза, характеризующаяся дефицитом красных кровяных пластинок - тромбоцитов, чаще вызванным иммунными механизмами. Признаками тромбоцитопенической пурпуры служат самопроизвольные, множественные, полиморфные кровоизлияния в кожу и слизистые оболочки, а также носовые, десневые, маточные и другие кровотечения. При подозрении на тромбоцитопеническую пурпуру оценивают анамнестические и клинические данные, показатели общего анализа крови, коагулограммы, ИФА, микроскопии мазков крови, пункции костного мозга. В лечебных целях больным назначаются кортикостероидные, гемостатические препараты, цитостатическая терапия, проводится спленэктомия.

Тромбоцитопеническая пурпура (болезнь Верльгофа, доброкачественная тромбоцитопения) - гематологическая патология, характеризующаяся количественным дефицитом тромбоцитов в крови, сопровождающаяся наклонностью к кровоточивости, развитию геморрагического синдрома. При тромбоцитопенической пурпуре уровень кровяных пластинок в периферической крови опускается значительно ниже физиологического - 150х10 9 /л при нормальном или несколько увеличенном количестве мегакариоцитов в костном мозге. По частоте встречаемости тромбоцитопеническая пурпура занимает первое место среди прочих геморрагических диатезов. Манифестирует заболевание обычно в детском возрасте (с пиком в раннем и дошкольном периоде). У подростков и взрослых патология в 2-3 раза чаще выявляется среди лиц женского пола.
В 45% случаев имеет место идиопатическая тромбоцитопеническая пурпура, развивающаяся самопроизвольно, без видимых причин. В 40% случаев тромбоцитопении предшествуют различные инфекционные заболевания (вирусные или бактериальные), перенесенные примерно за 2-3 недели до этого. В большинстве случаев это инфекции верхних отделов дыхательного тракта неспецифического генеза, в 20% - специфические (ветряная оспа, корь, краснуха, эпидемический паротит, инфекционный мононуклеоз, коклюш). Тромбоцитопеническая пурпура может осложнять течение малярии, брюшного тифа, лейшманиоза, септического эндокардита. Иногда тромбоцитопеническая пурпура проявляется на фоне иммунизации - активной (вакцинации) или пассивной (введения γ - глобулина). Тромбоцитопеническая пурпура может быть спровоцирована приемом медикаментов (барбитуратов, эстрогенов, препаратов мышьяка, ртути), длительным воздействием рентгеновских лучей (радиоактивных изотопов), обширным хирургическим вмешательством, травмой, избыточной инсоляцией. Отмечаются семейные случаи заболевания.
Большинство вариантов тромбоцитопенической пурпуры имеет иммунную природу и связано с продукцией антитромбоцитарных антител (IgG). Образование иммунных комплексов на поверхности тромбоцитов приводит к быстрому разрушению кровяных пластинок, уменьшению продолжительности их жизни до нескольких часов вместо 7-10 суток в норме.
Изоиммунная форма тромбоцитопенической пурпуры может быть обусловлена поступлением в кровь «чужеродных» тромбоцитов при повторных переливаниях крови или тромбоцитарной массы, а также антигенной несовместимостью тромбоцитов матери и плода. Гетероиммунная форма развивается при повреждении антигенной структуры тромбоцитов различными агентами (вирусами, медикаментами). Аутоиммунный вариант тромбоцитопенической пурпуры вызван появлением антител против собственных неизмененных антигенов тромбоцитов и обычно сочетается с другими заболеваниями такого же генеза (СКВ, аутоиммунной гемолитической анемией). Развитие трансиммунной тромбоцитопении у новорожденных провоцируется проходящими через плаценту антитромбоцитарными аутоантителами матери, больной тромбоцитопенической пурпурой.
Дефицит тромбоцитов при тромбоцитопенической пурпуре может быть связан с функциональным поражением мегакариоцитов, нарушением процесса отшнуровывания кровяных красных пластинок. Например, симптомокомплекс Верльгофа обусловлен неэффективностью гемопоэза при анемии (B-12 дефицитной, апластической), остром и хроническом лейкозах, системных заболеваниях органов кроветворения (ретикулезах), костномозговых метастазах злокачественных опухолей.
При тромбоцитопенической пурпуре происходит нарушение образования тромбопластина и серотонина, снижение сократительной способности и усиление проницаемости стенки капилляров. С этим связаны удлинение времени кровотечения, нарушение процессов тромбообразования и ретракции кровяного сгустка. При геморрагических обострениях количество тромбоцитов снижается вплоть до единичных клеток в препарате, в период ремиссии восстанавливается до уровня ниже нормы.
В классификации тромбоцитопенической пурпуры учитываются ее этиологические, патогенетические и клинические особенности. Различают несколько вариантов - идиопатическую (болезнь Верльгофа), изо-, транс-, гетеро- и аутоиммунную тромбоцитопеническую пурпуру, симптомокомплекс Верльгофа (симптоматическую тромбоцитопению).
По течению выделяют острую, хроническую и рецидивирующую формы. Острая форма более характерна для детского возраста, длится до 6 месяцев с нормализацией уровня тромбоцитов в крови, не имеет рецидивов. Хроническая форма протекает более 6 месяцев, чаще встречается у взрослых пациентов; рецидивирующая - имеет циклическое течение с повторениями эпизодов тромбоцитопении после нормализации уровня тромбоцитов.
Симптомы тромбоцитопенической пурпуры
Тромбоцитопеническая пурпура клинически проявляется при падении уровня тромбоцитов ниже 50х10 9 /л, обычно через 2-3 недели после воздействия этиологического фактора. Характерна кровоточивость по петехиально-пятнистому (синячковому) типу. У больных тромбоцитопенической пурпурой появляются безболезненные множественные кровоизлияния под кожу, в слизистые оболочки («сухой» вариант), а также кровотечения («влажный» вариант). Они развиваются спонтанно (часто в ночное время) и их выраженность не соответствует силе травматического воздействия.
Геморрагические высыпания полиморфны (от незначительных петехий и экхимозов до крупных синяков и кровоподтеков) и полихромны (от ярких багрово-синих до бледных желто-зеленых в зависимости от времени появления). Чаще всего геморрагии возникают на передней поверхности туловища и конечностей, редко - в области лица и шеи. Кровоизлияния определяются и на слизистой оболочке миндалин, мягкого и твердого неба, конъюнктиве и сетчатке, барабанной перепонке, в жировой клетчатке, паренхиматозных органах, серозных оболочках головного мозга.
Патогномоничны интенсивные кровотечения - носовые и десневые, кровотечения после удаления зубов и тонзиллэктомии. Могут появляться кровохарканье, кровавые рвота и понос, кровь в моче. У женщин обычно превалируют маточные кровотечения в виде меноррагий и метроррагий, а также овуляторных кровотечений в брюшную полость с симптомами внематочной беременности. Непосредственно перед менструацией появляются кожные геморрагические элементы, носовые и другие кровотечения. Температура тела остается в норме, возможна тахикардия. При тромбоцитопенической пурпуре имеется умеренная спленомегалия. При профузном кровотечении развивается малокровие внутренних органов, гиперплазия красного костного мозга и мегакариоцитов.
Медикаментозная форма манифестирует вскоре после приема лекарственного препарата, продолжается от 1 недели до 3 месяцев со спонтанным выздоровлением. Радиационная тромбоцитопеническая пурпура отличается тяжелым геморрагическим диатезом с переходом костного мозга в гипо- и апластическое состояние. Инфантильная форма (у детей до 2 лет) имеет острое начало, тяжелый, часто хронический характер и выраженную тромбоцитопению (9/л).
В течении тромбоцитопенической пурпуры выявляют периоды геморрагического криза, клинической и клинико-гематологической ремиссии. При геморрагическом кризе кровоточивость и лабораторные изменения ярко выражены, в период клинической ремиссии на фоне тромбоцитопении геморрагии не проявляются. При полной ремиссии отсутствуют и кровоточивость, и лабораторные сдвиги. При тромбоцитопенической пурпуре с большой кровопотерей наблюдается острая постгеморрагическая анемия, при длительной хронической форме - хроническая железодефицитная анемия.
Наиболее грозное осложнение - кровоизлияние в головной мозг развивается внезапно и быстро прогрессирует, сопровождаясь головокружением, головной болью, рвотой, судорогами, неврологическими нарушениями.

Диагноз тромбоцитопенической пурпуры устанавливается гематологом с учетом анамнеза, особенностей течения и результатов лабораторных исследований (клинического анализа крови и мочи, коагулограммы, ИФА, микроскопии мазков крови, пункции костного мозга).
На тромбоцитопеническую пурпуру указывают резкое снижение числа тромбоцитов в крови (9/л), увеличение времени кровотечения (>30 мин.), протромбинового времени и АЧТВ, снижение степени или отсутствие ретракции сгустка. Число лейкоцитов обычно в пределах нормы, анемия появляется при значительной кровопотере. На высоте геморрагического криза выявляются положительные эндотелиальные пробы (щипка, жгута, уколочная). В мазке крови определяется увеличение размеров и снижение зернистости тромбоцитов. В препаратах красного костного мозга обнаруживается нормальное или повышенное количество мегакариоцитов, присутствие незрелых форм, отшнуровка тромбоцитов в малочисленных точках. Аутоиммунный характер пурпуры подтверждается наличием в крови антитромбоцитарных антител.
Тромбоцитопеническую пурпуру дифференцируют от апластических или инфильтративных процессов костного мозга, острого лейкоза, тромбоцитопатий, СКВ, гемофилии, геморрагического васкулита, гипо- и дисфибриногенемий, ювенильных маточных кровотечений.
Лечение тромбоцитопенической пурпуры
При тромбоцитопенической пурпуре с изолированной тромбоцитопенией (тромбоциты >50х10 9 /л) без геморрагического синдрома лечение не проводится; при среднетяжелой тромбоцитопении (30-50 х10 9 /л) медикаментозная терапия показана в случае повышенного риска развития кровотечений (артериальной гипертензии, язвенной болезни желудка и 12-перстной кишки). При уровне тромбоцитов 9/л лечение осуществляют без дополнительных показаний в условиях стационара.
Кровотечения купируются введением кровоостанавливающих препаратов, местно применяется гемостатическая губка. Для сдерживания иммунных реакций и снижения сосудистой проницаемости назначаются кортикостероиды в понижающей дозе; гипериммунные глобулины. При больших кровопотерях возможны трансфузии плазмы и отмытых эритроцитов. Вливания тромбоцитарной массы при тромбоцитопенической пурпуре не показаны.
У больных хронической формой с рецидивами обильных кровотечений и кровоизлияниями в жизненно важные органы выполняют спленэктомию. Возможно назначение иммунодепрессантов (цитостатиков). Лечение тромбоцитопенической пурпуры при необходимости должно сочетаться с терапией основного заболевания.
Прогноз
В большинстве случаев прогноз тромбоцитопенической пурпуры весьма благоприятный, полное выздоровление возможно в 75% случаев (у детей - в 90%). Осложнения (например, геморрагический инсульт) наблюдаются в острой стадии, создавая риск смертельного исхода. При тромбоцитопенической пурпуре требуется постоянное наблюдение гематолога, исключаются препараты, влияющие на агрегационные свойства тромбоцитов (ацетилсалициловая к-та, кофеин, барбитураты), пищевые аллергены, проявляется осторожность при проведении вакцинации детей, ограничивается инсоляция.
Читайте также: