Влияние инсулина на обмен глюкозы в печени. Высвобождение глюкозы из печени
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
Углеводный обмен отвечает за процесс усвоения углеводов в организме, их расщепление с образованием промежуточных и конечных продуктов, а также новообразование из соединений, не являющихся углеводами, или превращение простых углеводов в более сложные. Основная роль углеводов определяется их энергетической функцией.
Глюкоза крови является непосредственным источником энергии в организме. Быстрота ее распада и окисления, а также возможность быстрого извлечения из депо обеспечивают экстренную мобилизацию энергетических ресурсов при стремительно нарастающих затратах энергии в случаях эмоционального возбуждения, при интенсивных мышечных нагрузках.
При снижении уровня глюкозы в крови развиваются:
вегетативные реакции (усиленное потоотделение, изменение просвета кожных сосудов).
Это состояние получило название «гипогликемическая кома». Введение в кровь глюкозы быстро устраняет данные расстройства.
Метаболизм углеводов в организме человека состоит из следующих процессов:
Расщепление в пищеварительном тракте поступающих с пищей поли- и дисахаридов до моносахаридов, дальнейшее всасывание моносахаридов из кишечника вкровь.
Синтез и распад гликогена в тканях (гликогенез и гликогенолиз).
Гликолиз (распад глюкозы).
Анаэробный путь прямого окисления глюкозы (пентозный цикл).
Анаэробный метаболизм пирувата.
Глюконеогенез — образование углеводов из неуглеводных продуктов.
Нарушения углеводного обмена
Всасывание углеводов нарушается при недостаточности амилолитических ферментов желудочно-кишечного тракта (амилаза панкреатического сока). При этом поступающие с пищей углеводы не расщепляются до моносахаридов и не всасываются. В результате у пациента развивается углеводное голодание.
Всасывание углеводов страдает также при нарушении фосфорилирования глюкозы в кишечной стенке, возникающем при воспалении кишечника, при отравлении ядами, блокирующими фермент гексокиназу (флоридзин, монойодацетат). Не происходит фосфорилирования глюкозы в кишечной стенке и она не поступает в кровь.
Всасывание углеводов особенно легко нарушается у детей грудного возраста, у которых еще не вполне сформировались пищеварительные ферменты и ферменты, обеспечивающие фосфорилирование и дефосфорилирование.
Причины нарушения углеводного обмена, вследствие нарушения гидролиза и всасывания углеводов:
нарушение функций печени - нарушение образования гликогена из молочной кислоты - ацидоз (гиперлакцидемия).
Нарушение синтеза и расщепления гликогена
Синтез гликогена может изменяться в сторону патологического усиления или снижения. Усиление распада гликогена происходит при возбуждении центральной нервной системы. Импульсы по симпатическим путям идут к депо гликогена (печень, мышцы) и активируют гликогенолиз и мобилизацию гликогена. Кроме того, в результате возбуждения центральной нервной системы повышается функция гипофиза, мозгового слоя надпочечников, щитовидной железы, гормоны которых стимулируют распад гликогена.
Повышение распада гликогена при одновременном увеличении потребления мышцами глюкозы происходит при тяжелой мышечной работе. Снижение синтеза гликогена происходит при воспалительных процессах в печени: гепатитах, в ходе которых нарушается ее гликоген-образовательная функция.
При недостатке гликогена тканевая энергетика переключается на жировой и белковый обмены. Образование энергии за счет окисления жира требует много кислорода; в противном случае в избытке накапливаются кетоновые тела и наступает интоксикация. Образование же энергии за счет белков ведет к потере пластического материала. Гликогеноз это нарушение обмена гликогена, сопровождающееся патологическим накоплением гликогена в органах.
Болезнь Гирке гликогеноз, обусловленный врожденным недостатком глюкозо-6-фосфатазы - фермента, содержащегося в клетках печени и почек.
Гликогеноз при врожденном дефиците α-глюкозидазы. Этот фермент отщепляет глюкозные остатки от молекул гликогена и расщепляет мальтозу. Он содержится в лизосомах и разобщен с фосфорилазой цитоплазмы.
При отсутствии α-глюкозидазы в лизосомах накапливается гликоген, который оттесняет цитоплазму, заполняет всю клетку и разрушает ее. Содержание глюкозы в крови нормальное. Гликоген накапливается в печени, почках, сердце. Обмен веществ в миокарде нарушается, сердце увеличивается в размерах. Больные дети рано умирают от сердечной недостаточности.
Нарушения промежуточного обмена углеводов
К нарушению промежуточного обмена углеводов могут привести:
Гипоксические состояния (например, при недостаточности дыхания или кровообращения, при анемиях), анаэробная фаза превращения углеводов преобладает над аэробной фазой. Происходит избыточное накопление в тканях и крови молочной и пировиноградной кислот. Содержание молочной кислоты в крови возрастает в несколько раз. Возникает ацидоз. Нарушаются ферментативные процессы. Снижается образование АТФ.
Расстройства функции печени, где в норме часть молочной кислоты ресинтезируется в глюкозу и гликоген. При поражении печени этот ресинтез нарушается. Развиваются гиперлакцидемия и ацидоз.
Гиповитаминоз В1. Нарушается окисление пировиноградной кислоты, так как витамин B1 входит в состав кофермента, участвующего в этом процессе. Пировиноградная кислота накапливается в избытке и частично переходит в молочную кислоту, содержание которой также возрастает. При нарушении окисления пировиноградной кислоты снижается синтез ацетилхолина и нарушается передача нервных импульсов. Уменьшается образование из пировиноградной кислоты ацетилкоэнзима А. Пировиноградная кислота является фармакологическим ядом для нервных окончаний. При увеличении ее концентрации в 2-3 раза возникают нарушения чувствительности, невриты, параличи и др.
При гиповитаминозе B1 нарушается также и пентозофосфатный путь обмена углеводов, в частности образование рибозы.
Гипергликемия
Гипергликемия это повышение уровня сахара крови выше нормального. В зависимости от этиологических факторов различают следующие виды гипергликемий:
Эмоциональная гипергликемия. При резком преобладании в коре головного мозга раздражительного процесса над тормозным возбуждение иррадиирует на нижележащие отделы центральной нервной системы. Поток импульсов по симпатическим путям, направляясь к печени, усиливает в ней распад гликогена и тормозит переход углеводов в жир. Одновременно возбуждение воздействует через гипоталамические центры и симпатическую нервную систему на надпочечники. Происходит выброс в кровь больших количеств адреналина, стимулирующего гликогенолиз.
Гормональные гипергликемии. Возникают при нарушении функции эндокринных желез, гормоны которых участвуют в регуляции углеводного обмена. Например, гипергликемия развивается при повышении продукции глюкагона - гормона α-клеток островков Лангерганса поджелудочной железы, который, активируя фосфорилазу печени, способствует гликогенолизу. Сходным действием обладает адреналин. К гипергликемии ведет избыток глюкокортикоидов (стимулируют глюконеогенез и тормозят гексокиназу) и соматотропного гормона гипофиза (тормозит синтез гликогена, способствует образованию ингибитора гексокиназы и активирует инсулиназу печени).
Гипергликемии при некоторых видах наркоза. При эфирном и морфинном наркозах происходит возбуждение симпатических центров и выход адреналина из надпочечников; при хлороформном наркозе к этому присоединяется нарушение гликогенообразовательной функции печени.
Гипергликемия при недостаточности инсулина является наиболее стойкой и выраженной. Ее воспроизводят в эксперименте путем удаления поджелудочной железы. Однако при этом дефицит инсулина сочетается с тяжелым расстройством пищеварения. Поэтому более совершенной экспериментальной моделью инсулиновой недостаточности является недостаточность, вызванная введением аллоксана (C4H2N2O4), который блокирует SH-группы. В β-клетках островков Лангерганса поджелудочной железы, где запасы SH-групп невелики, быстро наступает их дефицит и инсулин становится неактивным.
Экспериментальную недостаточность инсулина можно вызвать дитизоном, блокирующим цинк в β-клетках островков Лангерганса, что ведет к нарушению образования гранул из молекул инсулина и его депонирования. Кроме того, в β-клетках образуется дитизонат цинка, который повреждает молекулы инсулина.
Недостаточность инсулина может быть панкреатической и внепанкреатической. Оба эти вида инсулиновой недостаточности могут вызвать сахарный диабет.
Панкреатическая инсулиновая недостаточность
Этот тип недостаточности развивается при разрушении поджелудочной железы:
В этих случаях нарушаются все функции поджелудочной железы, в том числе и способность вырабатывать инсулин. После панкреатита в 16-18% случаев развивается инсулиновая недостаточность в связи с избыточным разрастанием соединительной ткани, которая нарушает снабжение клеток кислородом.
К инсулиновой недостаточности ведет местная гипоксия островков Лангерганса (атеросклероз, спазм сосудов), где в норме очень интенсивное кровообращение. При этом дисульфидные группы в инсулине переходят в сульфгидрильные и он не оказывает гипогликемического эффекта). Предполагают, что причиной инсулиновой недостаточности может послужить образование в организме при нарушении пуринового обмена аллоксана, близкого по структуре к мочевой кислоте.
Инсулярный аппарат может истощаться после предварительного повышения функции, например при излишнем употреблении в пищу легкоусвояемых углеводов, вызывающих гипергликемию, при переедании. В развитии панкреатической инсулиновой недостаточности важная роль принадлежит исходной наследственной неполноценности инсулярного аппарата.
Внепанкреатическая инсулиновая недостаточность
Этот тип недостаточности может развиться при повышенной активности инсулиназы: фермента, расщепляющего инсулин и образующегося в печени к началу полового созревания.
К недостаточности инсулина могут привести хронические воспалительные процессы, при которых в кровь поступает много протеолитических ферментов, разрушающих инсулин.
Избыток гидрокортизона, тормозящего гексокиназу, снижает действие инсулина. Активность инсулина снижается при избытке в крови неэстерифицированных жирных кислот, которые оказывают на него непосредственное тормозящее влияние.
Причиной недостаточности инсулина может послужить чрезмерно прочная его связь с переносящими белками в крови. Инсулин, связанный с белком, не активен в печени и мышцах, но оказывает обычно действие на жировую ткань.
В ряде случаев при сахарном диабете содержание инсулина в крови нормально или даже повышено. Предполагают, что диабет при этом обусловлен присутствием в крови антагониста инсулина, однако природа этого антагониста не установлена. Образование в организме антител против инсулина ведет к разрушению этого гормона.
Сахарный диабет
Углеводный обмен при сахарном диабете характеризуется следующими особенностями:
Резко снижен синтез глюкокиназы, которая при диабете почти полностью исчезает из печени, что ведет к уменьшению образования глюкозо-6-фосфата в клетках печени. Этот момент наряду со сниженным синтезом гликогенсинтетазы обусловливает резкое замедление синтеза гликогена. Происходит обеднение печени гликогеном. При недостатке глюкозо-6-фосфата тормозится пентозофосфатный цикл;
Активность глюкозо-6-фосфатазы резко возрастает, поэтому глюкозо-6-фосфат дефосфорилируется и поступает в кровь в виде глюкозы;
Тормозится переход глюкозы в жир;
Понижается прохождение глюкозы через клеточные мембраны, она плохо усваивается тканями;
Резко ускоряется глюконеогенез - образование глюкозы из лактата, пирувата, аминокислот жирных кислот и других продуктов неуглеводного обмена. Ускорение глюконеогенеза при сахарном диабете обусловлено отсутствием подавляющего влияния (супрессии) инсулина на ферменты, обеспечивающие глюконеогенез в клетках печени и почек: пируваткарбоксилазу, глюкозо-6-фосфатазу.
Таким образом, при сахарном диабете имеют место избыточная продукция и недостаточное использование глюкозы тканями, вследствие чего возникает гипергликемия. Содержание сахара в крови при тяжелых формах может достигать 4-5 г/л (400-500 мг%) и выше. При этом резко возрастает осмотическое давление крови, что ведет к обезвоживанию клеток организма. В связи с обезвоживанием глубоко нарушаются функции центральной нервной системы (гиперосмолярная кома).
Сахарная кривая при диабете по сравнению с таковой у здоровых значительно растянута во времени. Значение гипергликемии в патогенезе заболевания двояко. Она играет адаптивную роль, так как при ней тормозится распад гликогена и частично усиливается его синтез. При гипергликемии глюкоза лучше проникает в ткани и они не испытывают резкого недостатка углеводов. Гипергликемия имеет и отрицательное значение.
При ней повышается концентрация глюко- и мукопротеидов, которые легко выпадают в соединительной ткани, способствуя образованию гиалина. Поэтому для сахарного диабета характерно раннее поражение сосудов атеросклерозом. Атеросклеротический процесс захватывает коронарные сосуды сердца (коронарная недостаточность), сосуды почек (гломерулонефриты). В пожилом возрасте сахарный диабет может сочетаться с гипертонической болезнью.
Глюкозурия
В норме глюкоза содержится в провизорной моче. В канальцах она реабсорбируется в виде глюкозофосфата, для образования которого необходима гексокиназа, и после дефосфорилирования поступает в кровь. Таким образом, в окончательной моче сахара в нормальных условиях не содержится.
Суточный диурез возрастает до 5-10 л и более (полиурия). Развивается обезвоживание организм, развивается усиленная жажда (полидипсия). При нарушении углеводного обмена следует обратиться к эндокринологу за профессиональной помощью. Врач подберет необходимое медикаментозное лечение и разработает индивидуальную диету.
Поговорим про инсулин
Инсулин — это гормон поджелудочной железы, который главным образом воздействует на обмен веществ, причем в основном — на концентрацию глюкозы в крови. В своих тканях-мишенях он влияет как на мембранные, так и на внутриклеточные процессы. Некоторые из его эффектов перечислены в ниже.
Эффекты инсулина

Мембранные эффекты
- Стимуляция транспорта глюкозы (и некоторых других моносахаридов)
- Стимуляция транспорта аминокислот (особенно аргинина)
- Стимуляция транспорта жирных кислот
- Стимуляция поглощения клеткой К+ и Mg2+
Внутриклеточные эффекты
- Стимуляции синтеза РНК и ДНК
- Стимуляция синтеза белка
- Усиленная стимуляция гликогенсинтазы (гликогенез)
- Стимуляция глюкокиназы
- Ингибирование глюкозо-6-фосфатазы
- Стимуляция липогенеза
- Ингибирование липолиза (ингибирование синтеза цАМФ)
- Стимуляция синтеза жирных кислот
- Активация Mg2+-стимулируемой Na+/K+-АТФазы
Механизм действия инсулина и влияние его на обмен
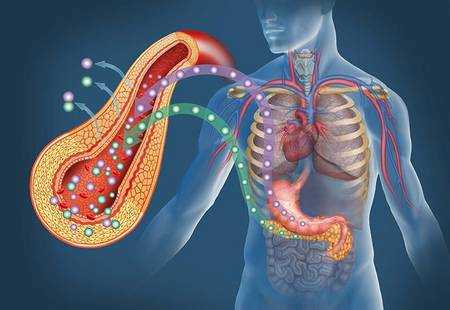
Инсулин и глюкоза
Попав в клетку, глюкоза быстро превращается в глюкозо-6-фосфат, поэтому ее внутриклеточная концентрация остается крайне низкой. Уровень глюкозы в артериальной крови в норме поддерживается в пределах 4-8 ммоль/л (72-144 мг/100 мл), так что по обе стороны клеточной мембраны всегда существует градиент ее концентраций. Несмотря на это, однако, простая диффузия обеспечивает поступление в большинство клеток лишь небольшого количества глюкозы, которого явно недостаточно для удовлетворения их метаболических потребностей (даже при возрастании концентрационного градиента, как это имеет место при высокой гипергликемии). В присутствии же инсулина проникновение декстрозы в клетки резко усиливается. Это действие инсулина проявляется лишь при наличии концентрационного градиента глюкозы, конкурентно ингибируется другими моносахаридами (например, галактозой) и следует кинетике насыщаемого процесса. Таким образом, гормон стимулирует процесс облегченной диффузии декстрозы, который осуществляется при участии чувствительных к гормону белковых транспортеров глюкозы (GLUT), расположенных на клеточной мембране. Эти транспортеры способны переносить глюкозу через клеточную мембрану в обоих направлениях, но ее поток зависит от концентрационного градиента, который направлен из внеклеточного пространства во внутриклеточное. В разных клетках найдены многочисленные GLUT, но инсулинозависимым является только один из этих белков — GLUT4, и именно он присутствует в мембранах клеток скелетных и сердечных мышц, а также жировой ткани.
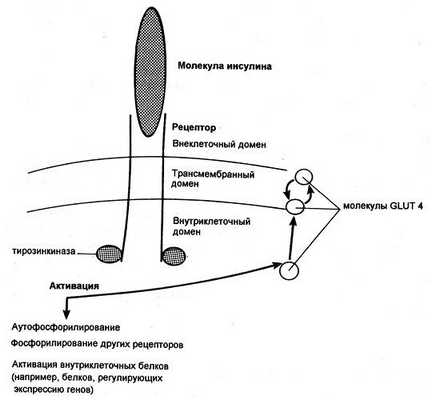
Димерный рецептор инсулина и последствия инсулиновой активации тирозинкиназы (GLUT — транспортер глюкозы)
Некоторые ткани полностью удовлетворяют свои потребности в глюкозе за счет инсулиннезависимых механизмов. Например, в клетки печени и центральной нервной системы декстроза попадает с помощью инсулиннезависимых GLUT, и поглощение этими тканями зависит только от ее уровня в крови. Кроме того, мембрану эритроцитов, клеток почек и кишечника глюкоза пересекает вместе с ионами натрия, которые поступают в клетки путем пассивной диффузии по градиенту концентрации.
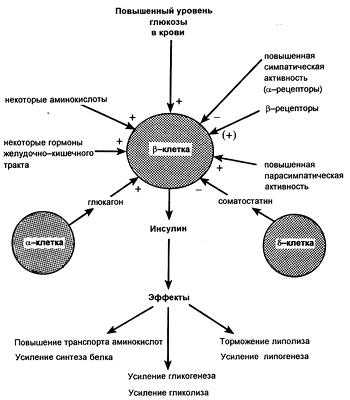
Регуляция продукции инсулина
Инсулин влияет и на внутриклеточные процессы обмена веществ. В печеночных и других клетках он стимулирует синтез гликогена, повышая активность гликогенсинтазы, что ускоряет включение гликозильных остатков в гликоген. Гормон поджелудочной железы повышает также активность печеночной глюкокиназы; этот фермент катализирует фосфорилирование глюкозы (с образованием глюкозо-6-фосфата). Одновременно гормон ингибирует печеночную фосфатазу, которая дефосфорилирует глюкозо-6-фосфат, с образованием свободной глюкозы. Такие изменения активности печеночных ферментов обусловливают снижение продукции декстрозы и наряду со стимуляцией поглощения ее периферическими клетками определяют гипогликемию, возникающее под влиянием инсулина. Возрастающая под действием последнего утилизация глюкозы в тканях обеспечивает сохранение запасов других внутриклеточных энергетических субстратов, таких как жиры и белки.
Белки и инсулин
Инсулин стимулирует не только активный транспорт аминокислот в периферические клетки, но и непосредственно синтез белка. Поскольку эти два эффекта могут не зависеть друг от друга, гормон влияет, очевидно, не только на клеточную мембрану, но и на внутриклеточные процессы. Стимуляции синтеза белка предшествует возрастание активности мРНК. Поскольку гормон с трудом проходит сквозь мембраны клеток, в механизме его ядерного эффекта должен принимать участие второй посредник. Синтез белка под действием инсулина усиливается и вследствие возрастания количества поступающих в клетку аминокислот. С другой стороны, возрастание утилизации глюкозы замедляет распад белка. Ускорение синтеза и замедление распада белка под влиянием гормона приводят к увеличению белковых запасов в интрацеллюлярном секторе.
Все эти эффекты определяют важнейшую роль инсулина в регуляции процессов роста и развития.
Инсулин и жир
Инсулин стимулирует поглощение и окисление глюкозы клетками жировой ткани. Он также стимулирует синтез липопротеиновой липазы в эндотелиальных клетках. Этот фермент катализирует гидролиз триглицеридов, связанных с липопротеинами крови, и способствует поступлению жирных кислот в адипоциты. Наряду с прямой стимуляцией липогенеза в печени и жировой ткани это приводит к увеличению запасов жира. Кроме того, инсулин ингибирует опосредуемый цАМФ липолиз, тормозя гормончувствительную внутриклеточную липопротеиновую липазу.
Инсулин и калий
Присутствие инсулина необходимо для поддержания внутриклеточной концентрации ионов калия; этот эффект, по всей вероятности, является следствием прямого влияния гормона на клеточную мембрану.
УЗ «Могилевская городская больница скорой медицинской помощи»
Основным источником энергии для клеток организма является глюкоза. Глюкоза образуется при всасывании углеводов, которые человек получает с пищей.
Когда человек не ест, нормальный уровень глюкозы крови поддерживается за счет использования запасов углеводов, которые есть в организме (гликоген печени) и синтеза глюкозы из белков. Часто спрашивают, если человек не ест, почему у него повышается глюкоза крови, откуда она берется. Ответ: из гликогена печени и распада белков. Однако запасов гликогена мало (примерно 90 грамм), а синтезировать глюкозу из белка организму крайне невыгодно, поэтому при голодании организм начинает «экономить» глюкозу и отключает ее поступление в часть органов. Т.е. при голодании глюкоза поступает только в критически важные органы (мозг, сосуды, почки, нервы).
Не пропускает глюкозу в ткани при голодании инсулин. Образно говоря, на клетках некоторых органов «висит замок», который открывается инсулином. Когда инсулин открывает замок, глюкоза поступает в клетку. Эти ткани являются инсулинозависимыми, глюкоза может попасть в них только тогда, когда инсулин «даст разрешение». Инсулинозависимыми тканями являются мышцы, жировая ткань.
Но в некоторые органы глюкоза попадает без инсулина, там нет замков, дверь для глюкозы всегда открыта. Эти органы называются инсулинНЕзависимыми. Смысл действия инсулина: есть еда, можем прокормить всех, инсулин открывает двери в клетки для глюкозы. Нет еды, значит, будем кормить только самые важные органы, инсулин закрывает двери для глюкозы в менее важных органах (можно оставить «голодными» мышцы и жир, но мозг нельзя).
Но что происходит, когда нет инсулина или он дефектный? Тогда глюкоза из углеводов пищи попадает в кровь, но не может поступить в ткани. Даже если уровень глюкозы в крови высокий, инсулинозависимые ткани голодают, дверь для глюкозы в клетке закрыта на замок, ключа нет или он сломан («голод среди изобилия»).
И в то же время в инсулинНЕзависимые ткани глюкоза поступает в излишних количествах. А что излишне, то нездорово. Глюкоза начинает связываться с белками этих тканей и повреждать их. Именно из-за этого при диабете и происходит повреждение органов-мишеней (нервов, сосудов, почек и др.).
Как происходит секреция инсулина в норме?
Минимальное количество инсулина в организме вырабатывается всегда (это называется базальная секреция инсулина). Когда человек поел, всасываются углеводы, в кровь поступает глюкоза, происходит выброс инсулина (это называется пищевой или прандиальный пик), двери открываются, глюкоза идет в клетку.
В норме секреция инсулина в ответ на прием пищи происходит в 2 фазы: первую быструю фазу (1-3 минуты) и вторую медленную (до 25-30 минут).
Сахарный диабет и хронические заболевания печени (Часть 2)
Сахарный диабет (СД) и хронические заболевания печени (ХЗП) — патологические состояния, ассоциированные друг с другом и достигающие масштабов эпидемии. Существует сильная патогенетическая взаимосвязь нарушений углеводного обмена и ряда ХЗП. Описаны единые механизмы, провоцирующие метаболические и аутоиммунные нарушения при развитии различных ХЗП, приводящие к стеатозу, инсулинорезистентности (ИР), нарушению толерантности к глюкозе и развитию СД. Эффективный контроль гликемии может оказать благоприятное влияние на лечение этих пациентов, и наоборот — имеются данные о положительном влиянии терапии ХЗП на углеводный обмен. Рассматриваются вопросы коррекции углеводного обмена у пациентов с ХЗП, приведены основные группы современных сахароснижающих препаратов, механизмы их действия, влияние на физиологию печени, возможности использования каждой из этих фармакологических групп у пациентов с нарушенной функцией печени. Перечислены современные подходы и возможности медикаментозного воздействия на процесс фиброгенеза при ХЗП, влияние этих препаратов на углеводный обмен. Ключевые слова: сахарный диабет, хронические заболевания печени, печеночная недостаточность, цирроз печени, вирусные гепатиты, алкогольная болезнь печени, неалкогольная жировая болезнь печени, сахароснижающие препараты.
Ключевые слова: сахарный диабет, хронические заболевания печени, печеночная недостаточность, цирроз печени, вирусные гепатиты, алкогольная болезнь печени, неалкогольная жировая болезнь печени, инсулинорезистентность.
- АБП — алкогольная болезнь печени
- АЛТ — аланинаминотрансфераза
- АСТ — аспартатаминотрансфераза
- ВГН — верхняя граница нормы
- ГПП — глюкагоноподобный пептид
- ИР — инсулинорезистентность
- ИФН — интерферон
- ЛС — лекарственные средства
- НАЖБП — неалкогольная жировая болезнь печени
- ПН — печеночная недостаточность
- ПТСД — посттрансплантационный сахарный диабет
- СД2 — сахарный диабет 2-го типа
- ТЗД — тиазолидиндионы
- ХЗП — хронические заболевания печени
- ЦП — цирроз печени
- CYP450 — цитохром P-450
- HbA1c — гликированный гемоглобин
Печень является главным органом метаболизма большинства лекарственных средств (ЛС). При хронических заболеваниях печени (ХЗП) отмечается снижение активности печеночных ферментов, метаболизирующих ЛС, что ведет к изменению фармакокинетики и фармакодинамики ЛС. Биодоступность ЛС, т. е количество поступившего в системную циркуляцию препарата по отношению к его введенной дозе, может увеличиваться или уменьшаться, соответствующим образом влияя на эффективность и токсичность ЛС. Это возможно в следующих случаях.
Увеличение биодоступности ЛС происходит при:
- повреждении и/или уменьшении количества гепатоцитов;
- ишемии гепатоцитов вследствие порто-системного шунтирования при циррозе печени (ЦП);
- снижении секреторной функции печени, что ведет к гипопротеинемии, в результате чего уменьшается связывание ЛС с белками, что может увеличивать концентрацию циркулирующих в крови фармакологически активных веществ.
Уменьшение биодоступности ЛС происходит в том случае, когда ЛС в результате распределения попадают в асцитическую или отечную жидкость и, таким образом, покидают системную циркуляцию и теряют активность [1].
Все это может привести к нежелательным побочным явлениям, в частности, при применении сахароснижающей терапии, увеличить риск возникновения гипогликемий или лактатацидоза.
Особенности сахароснижающей терапии при ХЗП
Бигуаниды (метформин)
Метформин является препаратом первой линии терапии сахарного диабета 2-го типа (СД2). Его воздействие на углеводный обмен обусловлено способностью улучшать чувствительность к инсулину, снижать инсулинорезистентность (ИР) периферических тканей, подавлять избыточную
продукцию глюкозы печенью и замедлять ее всасывание в кишечнике [2, 3].
Лактатацидоз — наиболее тяжелый возможный побочный эффект при применении бигуанидов, который развивается в результате неадекватной утилизации лактата путем его окисления или включения в глюконеогенез. Исследования последних лет продемонстрировали безопасность метформина в отношении развития лактатацидоза при условии строгого учета противопоказаний [3]. Одним из таковых, согласно последним клиническим рекомендациям ведущих медицинских организаций США, Европы и России, является печеночная недостаточность (ПН), однако степень ее по классификации Чайлд-Пью (табл. 1) [4] не уточняется.
Вопрос возможности назначения метформина при патологии печени остается предметом дискуссий и многих исследований. В 2014 г. опубликованы результаты наблюдательного ретроспективного исследования X. Zhang и соавт. о влиянии метформина на выживаемость пациентов с СД после установки диагноза ЦП. Медиана выживаемости выше у пациентов, получавших препарат (11,8 против 5,6 года в целом, р
Случаев развития лактатацидоза не наблюдалось [5]. Согласно анализу, проведенному R. Khan и соавт. [6], риск развития лактатацидоза у пациентов с повреждениями печени возникает при наличии множественной сопутствующей патологии, ведущей к тяжелой гипоксии. По мнению авторов, рекомендовано уменьшение максимальной суточной дозы препарата до 1500 мг с отменой при ухудшении печеночной и/или почечной функции. Метформин также оказал позитивное влияние на прогноз у пациентов с ЦП как исходом вирусного гепатита С, продемонстрировал снижение заболеваемости ЦП и ассоциированной с ним летальности [8].
Опубликован систематический обзор баз данных MEDLINE PubMed и Ovid MEDLINE за период с 2000 г. по март 2015 г. о применении метформина при различных патологиях печени: неалкогольная жировая болезнь печени (НАЖБП), HCV-, HBV-инфекциях, холангиокарциноме,
ЦП. Применение метформина ассоциировалось с увеличением выживаемости пациентов с ЦП различной этиологии.
Авторами выдвинуто предположение о возможности использования метформина в качестве терапии ХЗП, независимо от наличия нарушений углеводного обмена [9].
Тиазолидиндионы (ТЗД)
Основной терапевтический эффект ТЗД заключается в увеличении чувствительности периферических тканей (мышечной и жировой) к инсулину и как следствие — улучшении утилизации глюкозы. Группа ТЗД изначально включала в себя три препарата: троглитазон, росиглитазон, пиоглитазон, однако в связи с развитием тяжелых побочных эффектов при применении первых двух на сегодняшний день единственным представителем ТЗД, использующимся в клинической практике, является пиоглитазон. Гепатотоксичность пиоглитазона оценивалась в ходе многих клинических исследований, одно из которых проведено в Японии и включало более 20 тыс. пациентов с СД2. Случаев повышения активности печеночных трансаминаз не отмечалось [10]. Это находит подтверждение в недавно опубликованных результатах проспективного исследования F. Bril и соавт., которые тоже оценивали эффективность пиоглитазона у пациентов как с СД, так и без него [11].
R. Khan и соавт. рекомендуют ограничить максимальную суточную дозу пиоглитазона 30 мг [6]. Согласно официальной инструкции, применение препарата противопоказано при уровне аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (АСТ) более чем в 2,5 раза выше верхней границы нормы (ВГН) [12].
Средства, стимулирующие секрецию инсулина
К данному классу относят препараты сульфонилмочевины и глиниды. Они занимают лидирующее место в лечении СД2. Сахароснижающий эффект заключается в стимуляции секреции инсулина при условии сохранной функциональной активности β-клеток и зависит от уровня глюкозы плазмы,
в присутствии которой их стимулирующее влияние на секрецию и высвобождение инсулина усиливается. Элиминация препаратов данных фармакогрупп из организма осуществляется путем их биотрансформации в печени с участием изоферментов цитохрома P-450 (CYP450), метаболиты могут экскретироваться с мочой и желчью [2, 3]. Учитывая метаболизм данных ЛС в печени, у пациентов с нарушением ее функции фармакокинетика препаратов меняется: нарушается инактивация в печени, что приводит к повышению содержания ЛС в плазме, увеличению времени его полураспада и, соответственно, более высокому риску гипогликемии.
Представлены результаты лишь единичных исследований возможности применения секретагогов у пациентов с патологией печени. Препаратами выбора у пациентов с ХЗП без выявленной ПН являются ЛС с коротким периодом полураспада, такие как глипизид, натеглинид, репаглинид [13], при выявлении ПН рекомендовано уменьшить дозу в 2 раза. Назначение препаратов сульфонилмочевины и глинидов не рекомендовано пациентам с тяжелой ПН [6].
Ингбиторы α-глюкозидаз
К этой группе препаратов относятся средства, конкурентно ингибирующие ферменты желудочно-кишечного тракта и, в результате, замедляющие расщепление углеводов и поступление глюкозы в кровяное русло. Предотвращение быстрого поглощения глюкозы в проксимальном отделе тонкого кишечника приводит к уменьшению постпрандиальной гипергликемии [2, 3].
Результаты специальных клинических исследований показали безопасность применения акарбозы у пациентов с патологией печени. Так, данные рандомизированного двойного слепого исследования, включающего 100 пациентов с компенсированным ЦП и СД2, продемонстрировали улучшение показателей суточной гликемии [14]. Результаты другого плацебо-контролируемого исследования также показали возможность назначения препарата данной группы пациентам с тяжелой ПН, декомпенсированным ЦП и печеночной энефалопатией [15]. Таким образом, учитывая внепеченочный метаболизм, низкий риск развития гипогликемий, а также принимая во внимание опубликованные данные, назначение пациентам с ПН препаратов группы ингибиторов
α-глюкозидаз возможно.
Инсулины
В клинической практике инсулинотерапия, пожалуй, нашла самое широкое применение в сравнении с другими группами сахароснижающих препаратов у пациентов с патологией печени.
В целом, инсулинотерапия действительно является безопасной и эффективной антигипергликемической терапией, но при ее назначении следует принимать во внимание то, что вследствие дисфункции печени как главного органа, участвующего в метаболизме инсулина, потребность в нем может варьировать: она может как снижаться в результате угнетения глюконеогенеза, так и увеличиваться при нарастании ИР [6, 16]. Это затрудняет подбор оптимальных доз инсулинотерапии данной группе пациентов, определяет необходимость в частом определении уровня гликемии и коррекции терапии.
В настоящее время идет активное изучение изменения фармакокинетики различных инсулинов у пациентов с ПН.
Полученные данные позволяют отметить предпочтительность аналогов инсулина человека [17, 18]. Согласно действующим клиническим рекомендациям ведущих медицинских организаций США, Европы и России, противопоказаний и ограничений в назначении инсулинотерапии нет. Однако, принимая во внимание высокий риск развития гипогликемий и сложность коррекции инсулинотерапии, пациентам с ПН рекомендована редукция дозы инсулина на 25% под контролем показателей гликемии в течение суток [6, 7]. Особую осторожность следует проявлять в отношении пациентов, абстинентных к алкоголю [3, 6, 7, 19].
Препараты инкретинового ряда
Инкретины (англ. INtestinе, seCRETion, INsulin) — гормоны желудочно-кишечного тракта, стимулирующие секрецию инсулина в ответ на прием пищи. Наиболее изученными
являются глюкагоноподобный пептид-1 (ГПП-1) и глюкозозависимый инсулинотропный пептид. С точки зрения возможного применения у больных с СД2 наибольший интерес представляет ГПП-1 [19].
Результаты проведенного A. J. Scheen метаанализа клинических исследований изменения уровней печеночных трансаминаз при назначении как агонистов рецепторов ГПП-1, так и ингибиторов дипептидилпептидазы-4 — фермента, разрушающего ГПП-1, — пациентам с СД и ПН не показали существенных изменений уровня ферментов печени при применении этих ЛС по отдельности или в комбинации с другими сахароснижающими препаратами [20]. B. Giorda и соавт. отметили, что большинство мер предосторожности лишь отражают недостаток знаний об эффективности и безопасности инкретинов при ЦП [21]. На сегодняшний день в официальной инструкции лишь одного препарата данной фармакогруппы — вилдаглиптина — есть рекомендации относительно применения у пациентов с СД и ПН. Перед его назначением, а также регулярно в ходе первого года лечения (1 раз в 3 мес) рекомендуется определять уровень печеночных трансаминаз. При их повышении результат следует подтвердить повторным исследованием, а затем регулярно проводить определение биохимических показателей функции печени до тех пор, пока они не нормализуются. В случае подтверждения превышения активности АСТ или АЛТ до 3 ВГН препарат рекомендуется отменить [12].
Таким образом, предварительные данные свидетельствуют о безопасности применения препаратов инкретинового ряда в качестве антигипергликемической терапии у пациентов с ХЗП. Следует проявлять осторожность у пациентов с тяжелым ЦП по причине отсутствия достаточного клинического опыта [6, 7, 20].
Ингибиторы натрий-глюкозного котранспортера-2 (глифлозины)
Глифлозины ингибируют натрий-глюкозный котранспортер-2, что вызывает уменьшение реабсорбции натрия и глюкозы из просвета проксимального почечного канальца и приводит к развитию глюкозурии [2]. Свою безопасность глифлозины показали и в отношении пациентов с ПН. Опубликованы результаты исследования дапаглифлозина, канаглифлозина, эмпаглифлозина в отношении пациентов с ПН. Клинически значимых изменений фармакокинетических параметров у пациентов с СД2 и легкой и умеренной ПН не наблюдалось [19].
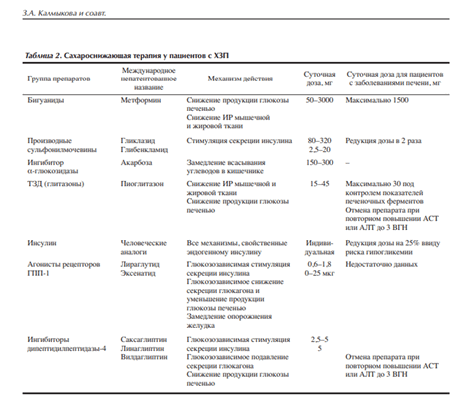
В табл. 2 представлены сводные данные по коррекции доз препаратов основных классов сахароснижающих средств у пациентов с заболеваниями печени [6, 12, 22].
Неотложные состояния при сахарном диабете

Неотложные состояния при сахарном диабете (СД) различаются по этиологии и патогенезу.
Выделяют следующие клинико-метаболические варианты острых осложнений в диабетологии:
- диабетический кетоацидоз и кетоацидотическая кома,
- гиперосмолярная кома и гиперосмолярное гипергликемическое состояние,
- молочнокислый ацидоз (лактат-ацидоз),
- гипогликемия и гипогликемическая кома.
ДИАБЕТИЧЕСКИЙ КЕТОАЦИДОЗ И КЕТОАЦИДОТИЧЕСКАЯ КОМА
Диабетический кетоацидоз (ДКА) - это критическое, ургентное состояние при СД, которое развивается вследствие абсолютного дефицита инсулина или выраженной относительной инсулиновой недостаточности, при несвоевременной диагностике и лечении, приводящее к развитию кетоацидотической комы с тяжелыми гормонально-метаболическими нарушениями органов и систем. ДКА, как правило, развивается при декомпенсации СД 1 типа, но вместе с тем редко может развиваться и при СД 2 типа. Это имеет подтверждение в исследованиях отечественных и зарубежных авторов (В. В. Потемкин, 2008, А. М. Мкртумян, 2008).
Причиной ДКА является абсолютный дефицит инсулина. Той или иной выраженности ДКА определяется у большинства пациентов на момент манифестации СД-1 (10—20 % всех случаев ДКА).
У пациента с установленным диагнозом СД-1 ДКА может развиться при прекращении введения инсулина, зачастую самим пациентом (13 % случаев ДКА), на фоне сопутствующих заболеваний, в первую очередь, инфекционных, при отсутствии увеличения дозы инсулина (30-40 %).
До 20 % случаев развития ДКА у молодых пациентов с СД-1 связаны с психологическими проблемами и/или нарушениями пищевого поведения (боязнь прибавки веса, боязнь гипогликемии, подростковые проблемы). Достаточно частой причиной ДКА в ряде стран является отмена инсулина самим пациентом из-за дороговизны препаратов для некоторых слоев населения (табл. 7. 11).
Вследствие нарастания кетонемии появляется тошнота, рвота, анорексия. Дефицит инсулина и повышенная секреция контринсулярных гормонов способствует распаду белков (катаболизм - протеолиз) в печени и образованию из них глюкозы в реакциях глюконеогенеза, а также аммиака, мочевины, что приводит к азотемии. Гипергликемия, гиперкетонемия, гиперазотемия приводят к нарушениям водно-электролитного обмена, повышению осмотического диуреза, выведению натрия, калия, фосфора, хлора. При полиурии вначале преобладает выделение натрия, поскольку он содержится во внеклеточной жидкости, а затем позднее присоединяется выход калия из клеток и повышенное его выделение с мочой. Развивается выраженное обезвоживание организма, уменьшается объём циркулирующей крови.
Таким образом, при кетоацидотической коме происходят глубокие метаболические нарушения, декомпенсация углеводного, липидного, белкового, электролитного обмена.
Токсическое воздействие кетоновых тел на клетки центральной нервной системы, угнетение ферментных систем, снижение утилизации глюкозы клетками мозга, кислородное голодание ведёт к нарушению сознания, развитию кетоацидотической комы.
Развитие ДКА в зависимости от вызвавшей его причины может занимать от нескольких недель до суток. В большинстве случаев ДКА предшествуют симптомы декомпенсации диабета, но иногда они могут не успеть развиться. Клинические симптомы ДКА включают полиурию, полидипсию, похудение, разлитые боли в животе («диабетический псевдоперитонит»), дегидратацию, выраженную слабость, запах ацетона изо рта (или фруктовый запах), постепенное помутнение сознания. Истинная кома при ДКА в последнее время в силу ранней диагностики развивается относительно редко. При физикальном обследовании выявляются признаки обезвоживания, тургора кожи и плотности глазных яблок, тахикардия, гипотония. В далеко зашедших случаях развивается дыхание Куссмауля. Более чем у 25% пациентов с ДКА развивается рвота, которая по цвету может напоминать кофейную гущу.
Базируется на данных клинической картины, указаниях на наличие у пациента СД-1, а также данных лабораторного исследования. Для ДКА характерна гипергликемия (в ряде случаев незначительная), кетонурия, метаболический ацидоз, гиперосмолярность.
Лабораторная диагностика острых осложнений сахарного диабета
Эффективная осмо-лярность, мОсм/кг
При обследовании пациентов с острой декомпенсацией СД необходимо определение уровня гликемии, креатинина и мочевины, электролитов, на основании чего производится расчет эффективной осмолярности. Кроме того, необходима оценка кислотно-основного состояния. Эффективная осмолярность (ЭО) рассчитывается по следующей формуле:
2 X [Na+ (мЭкв/л) + глюкоза (ммоль/л) ].
В норме ЭО составляет 285 - 295 мОсм/л.
У большинства пациентов с ДКА определяется лейкоцитоз, выраженность которого пропорциональна уровню кетоновых тел в крови. Уровень натрия, как правило, снижен вследствие осмотического оттока жидкости из интрацеллюлярных пространств в экстрацел-люлярные в ответ на гипергликемию. Реже уровень натрия может быть снижен ложноположительно как следствие выраженной гипертриглицеридемии. Уровень калия сыворотки исходно может быть повышен вследствие его перемещения из экстрацеллюлярных пространств.
Базируется на данных клинической картины, указаниях на наличие у пациента СД-1, а также данных лабораторного исследования. Для ДКА характерна гипергликемия (в ряде случаев незначительная), кетонурия, метаболический ацидоз, гиперосмолярность
При обследовании пациентов с острой декомпенсацией СД необходимо определение уровня гликемии, креатинина и мочевины, электролитов, на основании чего производится расчет эффективной осмолярности. Кроме того, необходима оценка кислотно-основного состояния. Эффективная осмолярность (ЭО) рассчитывается по следующей формуле: 2 X [Na+ (мЭкв/л) + глюкоза (ммоль/л) ]. В норме ЭО составляет 285 - 295 мОсм/л.
Другие причины потери сознания у пациентов с СД. Дифференциальная диагностика с гиперосмолярной комой, как правило, не вызывает затруднений (развивается у пожилых пациентов с СД-2) и не имеет большого клинического значения, т. к. принципы лечения обоих состояний сходны. При невозможности оперативно выяснить причину потери сознания пациента с СД ему показано введение глюкозы, т. к. гипогликемические состояния встречаются значительно чаще, а быстрая положительная динамика на фоне введения глюкозы сама по себе позволяет выяснить причину потери сознаний.
Лечение
Диабетический кетоацидоз, прекоматозное состояние и кома требуют немедленной госпитализации больного для проведения экстренной медицинской помощи. Необходимо срочное определение гликемии, глюкозурии, кетонемии и кетонурии, кислотно-щелочного равновесия, содержания натрия и калия, креатинина, мочевины, клинического анализа крови и мочи, ЭКГ, неврологическое обследование. На догоспитальном этапе или в приемном отделении после определения гликемии, глюкозурии, ацетонурии начинают внутривенно капельно инфузию 0, 9 % раствора хлорида натрия, при выраженной дегидратации до 1 л/час, инсулин короткого действия 20 ЕД в/м. Дальнейшее лечение осуществляют в реанимационном отделении или в отделении интенсивной терапии. Лечение ДКА легкой степени при сохраненном сознании и отсутствии тяжелой сопутствующей патологии можно проводить в эндокринологическом или терапевтическом отделении. В отделении реанимации и интенсивной терапии необходимо проводить мониторинг лабораторных показателей в целях предупреждения осложнений терапии - гипогликемии, гипокалиемии и гипонатриемии. Предлагаются следующие схемы лабораторного мониторинга:
Исследование глюкозы крови 1 раз в час, до снижения гликемии ниже 14 ммоль/л, затем 1 раз в 3 часа
Контроль ацетона мочи и кетоновых тел - 2 раза в первые 2 суток, затем 1 раз в сутки
Натрий и калий в плазме - не менее 2 раз в сутки
Фосфор - только у пациентов при недостаточности питания и хроническом алкоголизме
Остаточный азот, мочевина, креатинин сыворотки - исходно и через 3 дня
Гематокрит, газоанализ и рН - 1-2 раза в сутки до нормализации кислотно-основного состояния Терапия ДКА направлена на коррекцию основных патофизиологических нарушений. Основные компоненты лечебных мероприятий при кетоацидотической коме включают: устранение инсулиновой недостаточности, восстановление электролитного баланса и кислотно-основного равновесия, лечение сопутствующих заболеваний, которые могут быть причиной ДКА.
В настоящее время доказана целесообразность режима малых доз инсулина при лечении кетоацидотической комы, так как при введении больших доз инсулина имеется опасность развития гипогликемии, гипокалиемии, отёка мозга. Введение малых, физиологических доз инсулина проводится одновременно с регидратацией. При ДКА проводится внутривенная (в/в) инсулинотерапия в виде длительных инфузий. Для достижения оптимальных концентраций инсулина в крови необходима непрерывная инфузия малых доз инсулина - 0, 1 ЕД/кг/час, 4-10 ЕД инсулина в час (в среднем 6 ЕД/час). Это позволяет снизить липолиз, кетогенез и продукцию глюкозы печенью, таким образом воздействовать на главные звенья патогенеза ДКА. Начальная доза ИКД составляет 0, 15 ЕД/кг массы тела (в среднем 10-12 ЕД) и вводится в/в болюсно. Необходимую дозу инсулина набирают в инсулиновый шприц, добирают 0, 9 % NaCl до 1 мл вводят очень медленно (2-3 мин). Затем переходят на в/в введение ИКД по 0, 1 ЕД/кг/час (5-8 ЕД/час) с помощью инфузомата (первый вариант). Инфузионнную смесь готовят следующим образом: 50 ЕД ИКД+2 мл 20% раствора альбумина или 1 мл крови пациента для предотвращения сорбции инсулина в системе, и доводят общий объем до 50 мл 0, 9% NaCl. При отсутствии инфузомата используется второй вариант инсулинотерапии. С этой целью инфузионный раствор готовят из расчета 100 ЕД ИКД на каждые 100 мл 0, 9% раствора NaCl, концентрации ИКД будет составлять 1 ЕД/мл. Для предотвращения сорбции инсулина необходимо добавить 4 мл 20% альбумина на 100 мл раствора. Недостатком данного метода является трудность в титровании малых доз инсулина 12 по числу капель или мл смеси, а также возможность перегрузки жидкостью. Если инфузомата нет, более удобен в использовании 3 вариант. ИКД вводят в/в болюсно 1 раз в час шприцем в “резинку” инфузионной системы. Пример: в инсулиновый шприц набирают 6 ЕД ИКД, набирают в шприц 2 мл и добирают до 2 мл 0, 9% раствора NaCl и вводят медленно в течение 2-3 минут. Длительность фармакодинамического эффекта ИКД при этом составляет до 60 минут. Преимуществом этого способа введения является отсутствие сорбции инсулина, не нужно добавлять в раствор альбумин или кровь, а также более точен учет и коррекция введенной дозы ИКД. Внутримышечная (в/м) инсулинотерапия проводится при невозможности в/в доступа, в отсутствии нарушений гемодинамики и при легкой форме ДКА. Доза ИКД примерно 0, 4 ЕД/кг, из них половина вводится в/в, половина - в/м, затем введение ИКД осуществляется по 5-10 ЕД/час. Недостатками введения ИКД в/м является снижение его всасывания при нарушении микроциркуляции (коллапс, прекома, кома) и дискомфорт для больного (24 в/м инъекции в сутки). При этом, если через 2 часа после начала в/м введения ИКД гликемия не снижается, необходимо переходить на в/в введение. Скорость снижения гликемии при лечении ДКА должна быть не более 4 ммоль/час. Более резкое снижение гликемии создает опасность обратного осмотического градиента между внутри- и внеклеточным пространством, осмотического дисбаланса и отека мозга. В первые сутки не следует снижать гликемию ниже 13 ммоль/л. При отсутствии снижения гликемии в первые 2-3 часа следует провести коррекцию дозы инсулина - удвоить следующую дозу ИКД до 0, 2 ЕД/кг и проверить адекватность гидратации. Если снижение гликемии составило около 4 ммоль/л в час или достигнуто снижение гликемии до 14-15 ммоль/л, необходимо уменьшить следующую дозу ИКД вдвое (0, 05 ЕД/кг), примерно 2-4 ЕД/час. При снижении гликемии ниже 4 ммоль/час рекомендуется пропустить следующую дозу ИКД и продолжить ежечасно определять гликемию. После улучшения 13 состояния, стабилизации гемодинамики, уровень гликемии не более 11-12 ммоль/л и рН>7, 3 переходят на п/к введение ИКД каждые 4-6 часов с коррекцией дозы в зависимости от гликемии в сочетании с инсулином продленного действия 1-2 раза в сутки с начальной дозы 10-12 ЕД.
Адекватная регидратация восполняет не только водный и электролитный дефицит, но и приводит к снижению гликемии, улучшает периферическую гемодинамику и почечный кровоток. При исходно нормальном уровне натрия (менее 145 мэкв/л) для регидратации применяется изотонический (0, 9%) раствор хлорида натрия, при гипернатриемии (>150 мэкв/л) используют гипотонический раствор NaCl. Скорость регидратации составляет 1 литр в 1-й час (с учетом жидкости, введенной на догоспитальном этапе), затем по 0, 5 л во 2-й и 3-й час, по 0, 25-0, 5 л в последующие часы. Скорость регидратации корректируется в зависимости от показателей центрального венозного давления и клинической картины. Объем вводимой за час жидкости не должен превышать часовой диурез более чем на 500-1000 мл. Общее количество жидкости, введенной в первые 12 часов терапии, не должно превышать 10% массы тела.
При достижении уровня глюкозы крови 13-14 ммоль/л, переходят на введение 5-10% раствора глюкозы.
Восстановление электролитных нарушений
Важным разделом терапии кетоацитодической комы является коррекция электролитных нарушений. Развитие дефицита калия в организме, снижение его внутриклеточного содержания при кетоацитодической коме обусловлено повышенной экскрецией калия с потом, вследствие осмотического диуреза. Инсулиновая терапия, регидратация, снижение гликемии, уменьшение ацидоза способствуют поступлению калия в клетку вместе с глюкозой, в обмен на ионы водорода. Явления гипокалиемии проявляются обычно через 3-4 часа после начала инсулинотерапии и введения жидкости, при тенденции к нормолизации рН. Развитие гипокалиемии может приводить к тяжелым осложнениям со стороны сердечно-сосудистой системы - тахикардия, снижение АД, нарушения ритма; дыхательной системы, а также атонии желудка, кишечника, мочевого пузыря. Если уровень К+ плазмы неизвестен, в/в инфузию калия начинают не позднее, чем через 2 часа после начала инсулинотерапии, под контролем ЭКГ и диуреза. В/в инфузию калия начинают одновременно с введением инсулина по следующей схеме:
Читайте также: