Врожденная стационарная постоянная ночная слепота (синдром дисфункции палочек)
Добавил пользователь Alex Обновлено: 28.01.2026
• Врождённая стационарная ночная слепота с нормальной картиной глазного дна.
• Врождённая стационарная ночная слепота с патологическими изменениями на глазном дне — эта группа включает белоточечное глазное дно и болезнь Отучи
Белоточечное глазное дно
Определение
Белоточечпое глазное дно — состояние, при котором нарушен процесс регенерации зрительного пигмента, при этом постановление необходимого количества родонсина после воздействия яркого света происходит доныне, чем и норме.
Анамнез
Пациенты обратаются с жалобами на непрогресирующее нарушение
ночного зрении, при достаточной по времени адаптации зрение в темноте восстанавивается.
Эпидемиология и этиология
Генетика
Клинические признаки
Острота зрения. Обычно не изменяется.
Глазное дно. При исследовании глазного дна определяются множественные, жёлто-белые, мелкие точки в заднем полюсе, которые расходятся по направлению к периферии. Макула практически всегда не затронута (рис. 6-17, А-В).
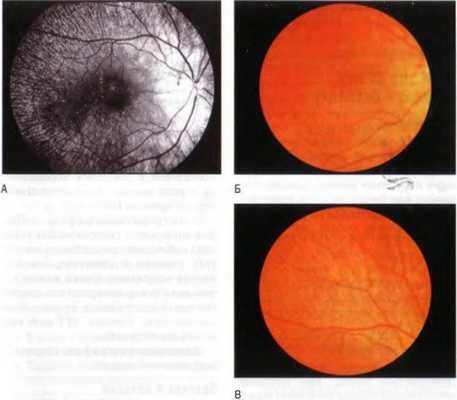
Рис. 6-17. Белоточечное глазное дно.
А. На фотографии в бескрасном свете видны радиально расходящиеся точки, распространяющиеся от заднего полюса к периферии сетчатки.
Б, В. На цветных фотографиях определяются множественные, жёлто-белые мелкие точки, расходящиеся от заднего полюса по направлению к периферии глазного дна. (Retina Slide Collection. Wills Eye Hospital. Philadelphia. Pennsylvania, compiled by Dr. Tamara Vrabec and Dr. Gordon Byrnes.)
Дифференциальная диагностика
Беспигментный ретинит (retinitis punctata albescens). Вариант пигментного ретинита, при котором на глазном дне обнаруживаются жёлто-белые точки, но при этом есть сужение сосудов и значительно сниженная ЭРГ, не восстанавливающаяся после продолжительной темновой адаптации.
Пятнистая сетчатка Кандори (Fleck retina of Kandori). Нарушение I с появлением крупных пятен и с менее тяжёлым нарушением ночного зрения.
Диагностика
Поля зрения. В норме. Темновая адаптометрия. Оба компонента адаптационной кривой -палочковый и колбочковый - очень медленно достигают конечного уровня.
Электроретинография. Важно знать, что при недостаточной темновой адаптации, а- и б-волны ЭРГ значительно снижены. Однако при более продолжительной темновой адаптации ЭРГ возвращается к нормальным показателям.
Электроокулография. Выявляют медленное восстановление светового подъёма при достаточной темновой адаптации.
Прогноз и лечение
Болезнь огучи (оguсhi's disease)
Болезнь Стучи вариант врождённой стационарной ночной слепоты, в основе которого лежит непрогрессирующее нарушение ночного зрения, связанное, как предполагается, с патологическим процессом фототрансдукции.
Зрительные пигменты фоторецепторов не изменены, а предположительным дефектом является нарушение фототрансдукции, что приводит к появлению патологических изменений ЭРГ.
Сетчатка имеет специфический серебристый блеск, при котором ретинальные сосуды отчётливо выделяются на фоне глазного дна. Такие изменения могут быть по всей сетчатке, только в заднем полюсе или только на периферии.
Феномен Мицуо-Накамура (mizuo-nakamura phenomenon). Металлический блеск сетчатки возникает после световой адаптации и исчезает через нескольких часов в темноте (рис. 6-18).
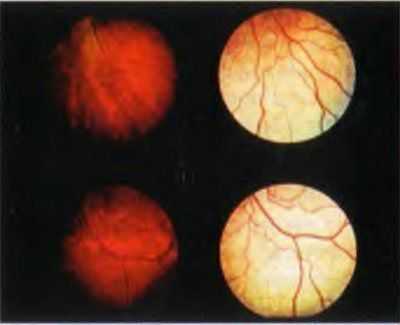
Рис. 6-18. Феномен Мицуо-Накамура. Металлический блеск сетчатки после воздействия света (фотографии справа) исчез после нескольких часов темновой адаптации (слева). (Retina Slide Collection. Wills Eye Hospital. Philadelphia. Pennsylvania, compiled by Dr. Tamara Vrahec and] Dr. Gordon Byrnes).
Поля зрения. Нормальные.
Темновая адаптация. Отмечают нормальную адаптацию колбочек, палочковая адаптация существенно замедленна и достигает нормального порога только через длительный период времени (от 3 до 24 ч).
Электроретинография. При фотопических и скотопических условиях наблюдают нормальную амплитуду а-волны и снижение б-волны или её отсутствие. Очень важно то, что даже после возвращения порога темновой адаптации к нормальным показателям, б-волна ЭРГ всё ещё может отсутствовать.
Электроокулография. Нормальный световой подъём.
Нарушение ночного зрения не прогрессирует. При болезни Огучи (Oguchi's Disease) эффективного лечения не существует.
Гемералопия
Гемералопия (куриная слепота) - офтальмопатология, характеризующаяся нарушением зрительной адаптации к условиям пониженной освещенности (сумеркам, темноте, искусственному затемнению). При гемералопии ухудшается видение предметов в темное время суток, нарушается пространственная ориентация в сумерках и процесс световой адаптации, сужаются поля зрения, возникают проблемы с цветовосприятием. Обследование пациентов с гемералопией включает визометрию, ахроматическую и цветовую периметрию, офтальмоскопию, биомикроскопию с линзой Гольдмана, адаптометрию, электроретинографию, оптическую когерентную томографию и др. Врожденная гемералопия неизлечима; при симптоматической форме проводится витаминотерапия, лечение первичной патологии сетчатки и зрительного нерва.
Общие сведения
Гемералопия (куриная или ночная слепота) характеризуется резким ухудшением зрения в темноте, сумерках, при смене освещения. Народное название гемералопии - «куриная слепота» произошло от схожести симптомов заболевания с особенностью зрения куриц, которые также плохо ориентируются в сумерках и темноте.
Сетчатка глаза содержит палочковидные («палочки») и колбочковидные («колбочки») светочувствительные клетки, которые образуют рецепторный аппарат глаза. Благодаря «палочкам» обеспечивается черно-белое ночное и сумеречное видение, с помощью «колбочек» - восприятие цветовой палитры днем. В среднем в сетчатке содержится около 110-125 млн. палочковидных и 6-7 млн. колбочковидных клеток (их нормальное соотношение составляет 18:1).
В палочковидных клетках сетчатки присутствует зрительный пигмент родопсин, обеспечивающий темновую адаптацию зрения. На свету родопсин распадается, а в темноте, при участии витамина А, восстанавливается. Синтез родопсина сопровождается выделением энергии, которая, преобразуясь в электрические импульсы, поступает по зрительному нерву в головной мозг. С помощью данного механизма обеспечивается нормальная деятельность палочковидных клеток и ночное зрение. При нарушении соотношения «палочек» и «колбочек» и недостатке родопсина развивается гемералопия - при слабом освещении острота зрения снижается, а при дневном ярком свете остается нормальной.
В офтальмологии выделяют три вида гемералопии: врожденную, симптоматическую и эссенциальную.
Причины гемералопии
Врожденная гемералопия обусловлена генетическими факторами и носит наследственно-семейный характер. Врожденная куриная слепота встречается при синдроме Ушера, наследственном пигментном ретините и другой наследуемой патологии.
Симптоматическая форма гемералопии развивается на фоне других заболеваний глаз: глаукомы, близорукости высоких степеней, ретинопатии, отслойки сетчатки, катаракты, хориоретинитов, атрофии зрительного нерва, сидероза, лучевых ожогов глаз (фотоофтальмии) и др.
Эссенциальная или функциональная гемералопия развивается при резком дефиците или отсутствии в организме витаминов А, В2, РР. Состояние гипо- и авитаминоза может встречаться при заболеваниях печени, малокровии, сильном истощении, сахарном диабете, лечении антагонистами ретинола (хинином), алкоголизме, заболеваниях ЖКТ с нарушением всасывания питательных веществ (хронический гастрит, энтерит, колит и др.).
Пусковыми факторами гемералопии могут выступать перенесенные инфекции (герпес, краснуха, корь, ветряная оспа), менопауза у женщин, диеты (в т. ч. вегетарианство). От появления гиповитаминоза до развития гемералопии может пройти около 2-х лет, так как в организме запасов витамина А хватает на год. Независимо от формы гемералопии, ухудшение зрения в темноте связано с одним и тем же механизмом - нарушением синтеза пигмента родопсина в палочковидных клетках сетчатки.
Симптомы гемералопии
Признаки врожденной гемералопии развиваются в раннем детстве: при этом стойкое снижение зрения не поддается излечению. Гемералопия сопровождается снижением остроты зрения в сумеречное и ночное время суток, чувством зрительного дискомфорта в полумраке. Человек с гемералопией замечает, что не различает окружающие предметы и теряет ориентацию в пространстве при слабом освещении, при переходе из хорошо освещенного помещения в темное. При этом днем и при достаточном освещении зрение, как правило, не нарушено.
Наблюдается ощущение «песка» и сухости в глазах. Дети с гемералопией боятся темноты, в связи с чем плачут и ведут себя беспокойно в сумерках. Гемералопия сопровождается сужением полей зрения и снижением восприятия желтого и синего цветов.
При эссенциальной гемералопии на конъюнктиве появляются ксеротические бляшки Искерского-Бито - плоские сухие пятна, расположенные в пределах глазной щели. Кроме глазных симптомов, отмечается сухость слизистых и кожных покровов, появление участков гиперкератоза на теле, шелушение и расчесы кожи, кровоточивость десен. При значительном дефиците витамина А может отмечаться размягчение и изъязвление роговицы (кератомаляция).
Диагностика гемералопии
При ухудшении сумеречного зрения необходимо проконсультироваться у офтальмолога, который поможет выявить возможные причины гемералопии. Обследование начинается с визометри - определения остроты зрения, которая при эссенциальной гемералопии часто бывает не изменена. Проведение ахроматической и цветовой периметрии позволяет выявить концентрическое сужение полей зрения, нарушение феномена Пуркинье.
Офтальмоскопическая картина при различных видах гемералопии имеет свои особенности. Так, при эссенциальной форме гемералопии глазное дно в норме, при других - имеет специфические изменения, характерные для заболевания, вызвавшего куриную слепоту. При врожденной гемералопии с помощью офтальмоскопии на сетчатке определяются мелкие округлые очаги дегенерации.
Для исследования темновой адаптации проводится адаптометрия. Функциональное состояние сетчатки оценивается с помощью электроретинографии и других дополнительных электрофизиологических исследований. Для выяснения причин симптоматической гемералопии показано выполнение тонографии, рефрактометрии, биомикроскопии с линзой Гольдмана, оптической когерентной томографии и т. д.
В комплексное обследование пациентов с гемералопией могут включаться консультации гастроэнтеролога, эндокринолога.
Лечение гемералопии
Врожденная форма гемералопии, ассоциированная с наследственной патологией, неизлечима - наблюдается устойчивое снижение сумеречного зрения. При приобретенных гемералопиях необходимо выяснение и устранение причин, приводящих к нарушению темновой адаптации.
При гемералопии, вызванной миопией, производится подбор очков или контактных линз, проводится лазерная коррекция близорукости, рефракционные операции (склеропластика, замена хрусталика и др.). Гемералопия, обусловленная глаукомой или катарактой, также требует хирургического лечения данных заболеваний (проведения антиглаукоматозных операций, экстракции или факоэмульсификации катаракты). При отслойке сетчатки показана лазерная коагуляция.
Эссенциальные гемералопии, в первую очередь, требуют нормализации питания: его обогащения продуктами, богатыми ретинолом и каротином (сливочным маслом, печенью трески, сыром, молоком, яичным желтком, морковью, шпинатом, томатами). Назначаются инстилляции витаминных глазных капель, прием витамина А, рибофлавина, никотиновой кислоты внутрь в возрастных дозировках. Одновременно необходимо лечение заболеваний ЖКТ, сахарного диабета (контроль уровня глюкозы крови, инсулинотерапия).
Прогноз и профилактика
Течение симптоматической гемералопии может привести как к восстановлению темновой зрительной адаптации, так и к стойкой утрате зрительной функции - прогноз определяется тяжестью основного заболевания. Функциональная гемералопия, как правило, хорошо поддается терапии и имеет благоприятный исход - полное восстановление сумеречного зрения. У пациентов с гемералопией нередко развивается патологическая боязнь темноты, которая может принимать характер фобии, невроза навязчивых состояний и психического расстройства.
Профилактика гемералопии требует обеспечения достаточного поступления в организм необходимых витаминов и защиты сетчатки глаза. С этой целью рекомендуется полноценное питание, использование защитных очков на солнце и при работе в условиях вредного излучения, лечение сопутствующей патологии. Лицам с гемералопией запрещается пользоваться лампами флюоресцентного свечения. Детям с легкой степенью миопии рекомендуется ношение очков в вечернее время.
Ночная слепота: причины, симптомы, диагностика, лечение
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Врожденная стационарная ночная слепота, или никталопия (отсутствие ночного зрения) - непрогрессирующее заболевание, причиной которого является дисфункция палочковой системы. При гистологическом исследовании структурных изменений в фоторецепторах не выявляют. Результаты электрофизиологических исследований подтверждают наличие первичного дефекта в наружном плексиформном (синаптическом) слое, так как нормальный палочковый сигнал не достигает биполярных клеток. Выделяют различные типы стационарной ночной слепоты, которые дифференцируются по ЭРГ.
Врожденная стационарная ночная слепота с нормальным глазным дном характеризуется разными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и сцепленным с Х-хромосомой.
Нормальное глазное дно
- Аутосомно-доминантная врожденная никталопия (тип Nugare): незначительная патология в колбочковой электроретинограммы и субнормальная палочковая электроретинограмма.
- Аутосомно-доминантная стационарная никталопия без миопии (тип Riggs): нормальная колбочковая электроретинограмма.
- Аутосомно-рецессивная или сцепленная с Х-хромосомой никталопия с миопией (тип Schubert- Bornschein).
Врожденная стационарная ночная слепота с изменением глазного дна. К этой форме заболевания относится болезнь Огуши - заболевание с аутосомно-рецессивным типом наследования, которое отличается от стационарной врожденной ночной слепоты изменениями на глазном дне, проявляющимися желтоватым металлическим блеском, более выраженным в заднем полюсе. Макулярная область и сосуды на этом фоне выглядят рельефно. После 3 ч темновой адаптации глазное дно становится нормальным (феномен Мицуо). После световой адаптации глазное дно вновь медленно приобретает металлический блеск. При исследовании темновой адаптации выявляют заметное удлинение палочкового порога при нормальной колбочковой адаптации. Концентрация и кинетика родопсина в норме.
С изменениями глазного дна
- Болезнь Огуши - аутосомно-рецессивное заболевание, характеризуется удлинением периода темновой адаптации до 2-12 ч для достижения нормальных палочковых порогов. Изменение цвета глазного дна с золотисто-коричневого цвета при световой адаптации до нормального в состоянии темповой адаптации (феномен Mizuo).
- «Белоточечное» глазное дно - аутосомно-рецессивное заболевание, характеризуется множественными мелкими бело-желтыми точками на заднем полюсе с интактной фовеа и распространением на периферию. Кровеносные сосуды, диск зрительного нерва, периферические поля и острота зрения остаются нормальными, электроретинограмма и электроокулограмма могут быть патологическими при рутинном исследовании и нормальными при длительной темповой адаптации.
Белоточечное глазное дно (fundus albi punctatus) сравнивают со звездным небом ночью, поскольку на средней периферии глазного дна и в макулярной области регулярно расположены мириады беловатых мелких нежных пятнышек. Заболевание с аутосомно-рецессивным типом наследования. На ФАГ выявляют фокальные области гиперфлюоресценции не связанные с белыми пятнами, которые на ангиограммах не видны.
В отличие от других форм стационарной ночной слепоты при белоточечном глазном дне отмечено замедление регенерации зрительного пигмента как в палочках, так и в колбочках. Амплитуда фотопических и скотопических а- и b-волн ЭРГ снижена при стандартных условиях регистрации. После нескольких часов темновой адаптации скотопический ответ ЭРГ медленно возвращается к норме.
Синдром Ушера ( Синдром Ашера )
Синдром Ушера - это редко встречающееся генетическое заболевание, протекающее с врожденной сенсоневральной тугоухостью, прогрессирующим пигментным ретинитом и вестибулярной атаксией. В зависимости от типа синдрома у пациентов присутствуют следующие признаки: значительная потеря слуха либо глухота, снижение зрения, нарушение равновесия, когнитивные расстройства. Диагностика включает офтальмологическое (визометрия, офтальмоскопия, электроретинография), отоневрологическое (аудиометрия, вестибулярные пробы), генетическое обследование. Лечение направлено на коррекцию слуха (слухопротезирование, КИ), поддержание зрения (вит. А, Е, омега-3).
МКБ-10
Синдром Ушера (синдром Ашера) - самая частая причина наследственной слепоглухоты. Его распространенность в популяции оценивается в 3,2-6,2 случая на 100 тыс. населения (по другим данным - 1:6000). Наибольшая заболеваемость отмечается среди евреев-ашкенази, франкоканадцев, испанских аргентинцев, финнов и др. «Первооткрывателем» заболевания считается германский офтальмолог А. фон Грефе. Однако свое «имя» синдром получил в честь британского окулиста Ч. Ушера, который в 1914 г. указал на наследственный характер болезни. Молекулярно-генетические механизмы синдрома были расшифрованы в 1995 г., что открыло широкие возможности для его изучения.
Причины
В настоящее время известно более десятка генов, дефекты которых могут привести к развитию синдрома Ушера. Все эти гены, несмотря на разную локализацию и функцию, входят в состав трансмембранного белкового комплекса, участвующего в перемещении миозина, функционировании фоторецепторов сетчатки, а также волосковых клеток улитки. Наиболее часто мутации обнаруживаются в следующих генах:
- CDH23 - «ген глухоты», кодирует белок кадгерин-23;
- MYO7A - ген миозина VIIA;
- PCDH15 - ген протокадгерина-15;
- USH1С - ген гармонина;
- USH1G - ген анкириноподобного белка;
- USH2A - ген ушерина;
- ADGRV1 - ген адгезии G-белка;
- CLRN1 - ген кларина-1 и др.
Синдром Ушера наследуется аутосомно-рецессивным путем от обоих родителей, являющихся носителями дефектных генов. Чаще болеют представители закрытых этнических групп, среди которых достаточно часты близкородственные браки.
Патогенез
Белковые продукты, кодируемые названными генами, принимают непосредственное участие в развитии и функционировании рецепторного аппарата сетчатки глаза и внутреннего уха. Генетические мутации приводят к нарушению формирования воспринимающего аппарата - фоторецепторов и волосковых клеток, что сопровождается врожденными или рано дебютирующими нарушениями зрительной, слуховой и вестибулярной функции.
При синдроме Ушера на глазном дне накапливаются гранулы пигмента, которые распространяются от центра к периферии. Со временем происходит сужение полей и снижение остроты зрения. Патология также затрагивает структуры внутреннего уха: отмечается атрофия спирального узла, нервных волокон кортиева органа, сосудистой полоски улитки.
Классификация
Синдром Ушера генетически неоднороден. Выделяют 4 клинических подтипа, различающихся молекулярно-генетическими механизмами, возрастом манифестации и выраженностью симптоматики:
- Тип 1. Сопряжен с мутациями в генах MYO7A, CDH23, PCDH15, USH1G, USH1C. Протекает наиболее тяжело. Характерна врожденная глухота или глубокая тугоухость. Вестибулярные нарушения и признаки пигментного ретинита развиваются до 5 лет. Составляет около 30% всех случаев.
- Тип 2. Вызывается мутациями USH2A, DFNB31, ADGRV1. Сопровождается непрогрессирующей тугоухостью, ухудшением зрения после 10 лет. Вестибулярная функция не нарушена. Диагностируется примерно у 60% пациентов.
- Тип 3. Связан с мутациями гена CLRN1. Протекает с постепенным ухудшением слуха и зрения, часто - с вестибулярной дисфункцией. Изменения сетчатки развиваются после 20 лет. Выявляется у 3% больных.
- Тип 4. Атипичный вариант синдрома Ушера. Обусловлен дефектами генов HARS, PDZD7, CEP250, C2orf71, может наследоваться Х-сцеплено.
Симптомы
Классическая клиническая картина развивается при 1-м типе синдрома Ушера. Уже в раннем детстве у ребенка диагностируется тяжелая сенсоневральная тугоухость или полная глухота. Наблюдается задержка психомоторного развития, дети поздно начинают ходить. В дошкольном возрасте обнаруживается зрительная дисфункция: в результате усугубления пигментного ретинита быстро ухудшается периферическое зрение, развивается ночная слепота (гемералопия). Это проявляется затруднением ориентации в темноте, частыми спотыканиями, столкновениями с препятствием (другой человек, дверной проем, мебель).
Возникают вестибулярные расстройства: головокружение, нарушение равновесия, атактическая походка. В некоторых случаях может отмечаться когнитивный дефицит, психозы. При других типах болезни Ушера зрительные и слуховые нарушения развиваются позднее и выражены в меньшей степени; вестибулярный синдром не отмечается.
Осложнения
Нейросенсорная тугоухость и постепенная утрата зрения приводят к глубокой инвалидизации. Дети с синдромом Ушера нуждаются в специальном обучении, психолого-педагогическом сопровождении, создании безопасной окружающей среды. Вестибулярные нарушения могут провоцировать падения, повышать риск травматизма. Вторично страдает речь и интеллектуальная сфера. Одним из частых осложнений является катаракта. Прогрессивное снижение зрения приводит к тому, что к 40-50 годам больные синдромом Ушера могут полностью ослепнуть. В тяжелых случаях возникает слепоглухота.
Поскольку нарушения при синдроме Ушера затрагивают несколько анатомо-функциональных систем, диагностика должна носить мультидисциплинарный подход. Больным необходимы консультации отоларинголога, офтальмолога, генетика, проведение комплексного инструментально-лабораторного обследования:
- Офтальмологический осмотр.Визометрия и осмотр глазного дна не всегда позволяют обнаружить начальные признаки пигментной дегенерации сетчатки. В этом отношении наиболее чувствительным тестом является электроретинография, выявляющая изменения еще на доклиническом уровне. По результатам периметрии отмечается концентрическое сужение полей зрения.
- Исследование слуха. Степень снижения слуха устанавливается с помощью аудиометрии. Дополнительно регистрируется отоакустическая эмиссия, стволовые ВП, проводится электрокохлеарография. Больного осматривает врач-сурдолог.
- Исследование вестибулярного анализатора. Для выявления вестибулярных нарушений выполняется видеонистагмография, вестибулометрические пробы.
- Генодиагностика. Проводится секвенирование образцов генетического материала пациента. На основе обнаруженных изменений определяется генетический вариант синдрома Ушера. Также необходимо медико-генетическое консультирование членов семьи.
Дифференциальная диагностика
Синдром Ушера необходимо отличать от других синдромальных и несиндромальных форм глухоты и слепоты, развивающихся при:
- врожденной краснухе - врожденная тугоухость, катаракта, ВПС;
- синдроме Альстрема - дегенерация сетчатки, сенсоневральная тугоухость, ожирение, СД, кардиомиопатия, нефропатия;
- синдроме Халлгрена (акустико-ретино-церебеллярной дегенерации) - пигментный ретинит, катаракта, тугоухость, мозжечковый синдром.
Лечение синдрома Ушера
Консервативная терапия
На сегодняшний день методов полного излечения заболевания не разработано. Все предпринимаемые меры направлены на компенсацию нарушенных функций и замедление течения патологии. Больным синдромом Ушера рекомендовано проведение ежегодных поддерживающих курсов медикаментозной терапии, включающей ноотропы, сосудорасширяющие препараты, антиоксиданты. С целью торможения прогрессирования пигментного ретинита рекомендуется прием больших доз ретинола пальмитата, соблюдение диеты, богатой содержанием витаминов А, Е, ПНЖК.
Способ коррекции слуховой функции подбирается индивидуально. Выбор может быть сделан в пользу слухового протезирования или установки кохлеарного импланта. Для адаптации детей с синдром Ушера к жизни в социуме важна помощь психологов, сурдопедагогов.
Экспериментальное лечение
Сообщается о разработке генной терапии для больных с синдромом Ушера 1 типа. Клинические испытания проходит препарат UshStat, который представляет собой лентивирусный вектор для доставки неповрежденного гена MYO7A. Предполагается субретинальное введение UshStat с целью восстановления функции зрения.
Качество жизни пациентов с синдромом Ушера значительно страдает из-за проблем со зрением и слухом. Около 50% лиц с 1-м и 70% со 2-м типом заболевания сохраняют остроту зрения от 20 до 40% на обоих или одном глазу. Больным необходимо пожизненное соблюдение диеты, защита глаз от ультрафиолета. Профилактика заключается в проведении генетических консультаций и лабораторного тестирования в семьях, где имеются случаи данного заболевания. Возможна пренатальная диагностика синдрома Ушера.
1. Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов/ Иванова М.Е , Атарщиков Д.С. , Демчинский А.М. , Стрельников В.В. , Бар Д. , Порядин Г.В. , Балашова Л.М. , Салмаси Ж.М.// РМЖ «Клиническая Офтальмология». - 2019. - №4.
3. Этиологические аспекты врожденной тугоухости/ Коноплев О.И. и др./ Медицинский вестник Северного Кавказа. - 2019.
Синдром WPW
Синдром Вольфа-Паркинсона-Уайта (синдром WPW) - клинико-электрокардиографический синдром, характеризующийся предвозбуждением желудочков по дополнительным атриовентрикулярным путям проведения и развитием пароксизмальных тахиаритмий. Синдром WPW сопровождается различными аритмиями: наджелудочковой тахикардией, фибрилляцией или трепетанием предсердий, предсердной и желудочковой экстрасистолией с соответствующей субъективной симптоматикой (ощущением сердцебиения, одышкой, гипотензией, головокружением, обмороками, болями в грудной клетке). Диагностика синдрома WPW основана на данных ЭКГ, суточного ЭКГ-мониторирования, ЭхоКГ, ЧПЭКС, ЭФИ. Лечение синдрома WPW может включать антиаритмическую терапию, чреспищеводную электрокардиостимуляцию, катетерную РЧА.
Синдром Вольфа-Паркинсона-Уайта (синдром WPW) - синдром преждевременного возбуждения желудочков, обусловленный проведением импульсов по добавочным аномальным проводящим пучкам, соединяющим предсердия и желудочки. Распространенность синдрома WPW, по данным кардиологии, составляет 0,15-2%. Синдром WPW чаще встречается среди мужчин; в большинстве случаев манифестирует в молодом возрасте (10-20 лет), реже - у лиц старшего возраста. Клиническое значение синдрома WPW заключается в том, что при его наличии часто развиваются тяжелые нарушения сердечного ритма, которые представляют угрозу для жизни больного и требуют особых подходов к лечению.
Причины синдрома WPW
По мнению большинства авторов, синдром WPW, обусловлен сохранением добавочных атриовентрикулярных соединений в результате незавершенного кардиогенеза. При этом происходит неполная регрессия мышечных волокон на этапе формирования фиброзных колец трикуспидального и митрального клапанов.
В норме дополнительные мышечные пути, соединяющие предсердия и желудочки, существуют у всех эмбрионов на ранних стадиях развития, но постепенно они истончаются, сокращаются и полностью исчезают после 20-й недели развития. При нарушении формирования фиброзных атриовентрикулярных колец мышечные волокна сохраняются и составляют анатомическую основу синдрома WPW. Несмотря на врожденный характер дополнительных АВ-соединений, синдром WPW может впервые проявиться в любом возрасте. При семейной форме синдрома WPW чаще имеют место множественные добавочные атриовентрикулярные соединения.
Классификация синдрома WPW
По рекомендации ВОЗ, различают феномен и синдром WPW. Феномен WPW характеризуется электрокардиографическими признаками проведения импульса по дополнительным соединениям и предвозбуждением желудочков, но без клинических проявлений АВ реципрокной тахикардии (re-entry). Под синдромом WPW подразумевается сочетание предвозбуждения желудочков с симптоматической тахикардией.
С учетом морфологического субстрата выделяют несколько анатомических вариантов синдрома WPW.
I. С добавочными мышечными АВ-волокнами:
- идущими через добавочное левое или правое париетальное АВ-соединение
- идущими через аортально-митральное фиброзное соединение
- идущими от ушка правого или левого предсердия
- связанными с аневризмой синуса Вальсальвы или средней вены сердца
- септальными, парасептальными верхними или нижними
II. Со специализированными мышечными АВ-волокнами («пучками Кента»), происходящими из рудиментарной, аналогичной структуре атриовентрикулярного узла, ткани:
- атрио-фасцикулярными - входящими в правую ножку пучка Гиса
- входящими в миокард правого желудочка.
Выделяют несколько клинических форм синдрома WPW:
- а) манифестирующую - с постоянным наличием дельта-волны, синусовым ритмом и эпизодами атриовентрикулярной реципрокной тахикардии.
- б) интермиттирующую - с преходящим предвозбуждением желудочков, синусовым ритмом и верифицированной атриовентрикулярной реципрокной тахикардией.
- в) скрытую - с ретроградным проведением по дополнительному атриовентрикулярному соединению. Электрокардиографические признаки синдрома WPW не выявляются, имеются эпизоды атриовентрикулярной реципрокной тахикардии.
Патогенез синдрома WPW
Синдром WPW обусловлен распространением возбуждения от предсердий к желудочкам по дополнительным аномальным путям проведения. В результате этого возбуждение части или всего миокарда желудочков происходит раньше, чем при распространении импульса обычным путем - по АВ-узлу, пучку и ветвям Гиса. Предвозбуждение желудочков отражается на электрокардиограмме в виде дополнительной волны деполяризации - дельта-волны. Интервал P-Q(R) при этом укорачивается, а длительность QRS увеличивается.
Когда в желудочки приходит основная волна деполяризации, их столкновение в сердечной мышце регистрируется в виде так называемого сливного комплекса QRS, который становится несколько деформированным и уширенным. Нетипичное возбуждение желудочков сопровождается нарушением последовательности процессов реполяризации, что находит выражение на ЭКГ в виде дискордантного комплексу QRS смещения RS-T сегмента и изменения полярности зубца T.
Возникновение при синдроме WPW пароксизмов суправентрикулярной тахикардии, мерцания и трепетания предсердий связано с формированием круговой волны возбуждения (re-entry). В этом случае импульс по AB-узлу движется в антероградном направлении (от предсердий к желудочкам), а по дополнительным путям - в ретроградном направлении (от желудочков к предсердиям).
Симптомы синдрома WPW
Клиническая манифестация синдрома WPW происходит в любом возрасте, до этого его течение может быть асимптомным. Синдром WPW сопровождается различными нарушениями сердечного ритма: реципрокной наджелудочковой тахикардией (80%), фибрилляцией предсердий (15-30%), трепетанием предсердий (5%) с частотой 280-320 уд. в мин. Иногда при синдроме WPW развиваются менее специфичные аритмии - предсердная и желудочковая экстрасистолия, желудочковая тахикардия.
Приступы аритмии могут возникать под влиянием эмоционального или физического перенапряжения, злоупотребления алкоголем или спонтанно, без видимых причин. Во время аритмического приступа появляются ощущения сердцебиения и замирания сердца, кардиалгии, чувство нехватки воздуха. Мерцание и трепетание предсердий сопровождается головокружением, обмороками, одышкой, артериальной гипотензией; при переходе в фибрилляцию желудочков может наступить внезапная сердечная смерть.
Пароксизмы аритмии при синдроме WPW могут длиться от нескольких секунд до нескольких часов; иногда они купируются самостоятельно или после выполнения рефлекторных приемов. Затяжные пароксизмы требуют госпитализации больного и вмешательства кардиолога.
Диагностика синдрома WPW
При подозрении на синдром WPW проводится комплексная клинико-иснтрументальная диагностика: ЭКГ в 12 отведениях, трансторакальная эхокардиография, мониторирование ЭКГ по Холтеру, чреспищеводная электрокардиостимуляция, электрофизиологическое исследование сердца.
К электрокардиографическим критериям синдрома WPW относятся: укорочение PQ-интервала (менее 0,12 с), деформированный сливной QRS-комплекс, наличие дельта-волны. Суточное ЭКГ мониторирование применяется для обнаружения преходящих нарушений ритма. При проведении УЗИ сердца выявляются сопутствующие пороки сердца, кардиомиопатию.
Проведение чреспищеводной электрокардиостимуляции при синдроме WPW позволяет доказать наличие дополнительных путей проведения, индуцировать пароксизмы аритмии. Эндокардиальное ЭФИ позволяет точно определить локализацию и количество дополнительных путей, верифицировать клиническую форму синдрома WPW, выбрать и оценить эффективность лекарственной терапии или РЧА. Дифференциальную диагностику синдрома WPW проводят с блокадами ножек пучка Гиса.
Лечение синдрома WPW
При отсутствии пароксизмов аритмии синдром WPW не требует специального лечения. При гемодинамически значимых приступах, сопровождающихся синкопэ, стенокардией, гипотензией, нарастанием признаков сердечной недостаточности, требуется выполнение незамедлительной наружной электрической кардиоверсии или чреспищеводной электрокардиостимуляции.
В некоторых случаях для купирования пароксизмов аритмий эффективными оказываются рефлекторные вагусные маневры (массаж каротидного синуса, проба Вальсальвы), внутривенное введение АТФ или блокаторов кальциевых каналов (верапамила), антиаритмических препаратов (новокаинамида, аймалина, пропафенона, амиодарона). В дальнейшем пациентам с синдромом WPW показана постоянная антиаритмическая терапия.
В случае резистентности к антиаритмическим препаратам, развития фибрилляцией предсердий проводится катетерная радиочастотная абляция добавочных путей проведения трансаортальным (ретроградным) или транссептальным доступом. Эффективность РЧА при синдроме WPW достигает 95%, риск рецидивов составляет 5-8 %.
Прогноз и профилактика синдрома WPW
У пациентов с бессимптомным течением синдрома WPW прогноз благоприятный. Лечение и наблюдение требуется только лицам, имеющим отягощенный семейный анамнез в отношении внезапной смерти и профессиональные показания (спортсменам, летчикам и др.). При наличии жалоб или жизнеугрожающих аритмий необходимо проведение полного комплекса диагностического обследования для выбора оптимального метода лечения.
Пациенты с синдромом WPW (в том числе, перенесшие РЧА) нуждаются в наблюдении кардиолога-аритмолога и кардиохирурга. Профилактика синдрома WPW носит вторичный характер и заключается в проведении противоаритмической терапии для предотвращения повторных эпизодов аритмий.
Читайте также:
- Лечение бактериального менингита у детей. Антибиотики
- Обрезание у мальчиков: показания, противопоказания, особенности
- Перелом головки мыщелка плечевой кости. Диагностика и лечение
- Гистиоцитарные заболевания глаза: причины, диагностика, лечение
- Закрытые повреждения таза. Закрытые повреждения позвоночника.