
Консервативные пептиды вирусных оболочек
За последние годы отмечается резкий рост и распространение инфекционных заболеваний вирусной этиологии, в частности ВИЧ-инфекции, вирусных гепатитов, герпеса и др. Поэтому создание эффективных противовирусных вакцин и препаратов становится сейчас одной из важнейших проблем.
С использованием методов протеомики и биоинформатики в НИИ биомедицинской химии им. В.Н. Ореховича (г. Москва) занимаются созданием вакцин против вируса гепатита С. Проводился компьютерный анализ строения белков вируса гепатита С. Была создана база данных, в этой базе данных были выявлены участки полипептидной цепи белковых молекул, которые, по сути дела, инвариантны (идентичны) для всех 827 последовательностей оболочечных белков вируса гепатита С. Эти участки получили название консервативные пептиды, а их открытие создало новые возможности для получения противовирусных вакцин. Известно, что постоянная изменчивость вирусных белков - основное препятствие для создания таких вакцин, а найденные участки консервативны и не подвержены мутациям. Поэтому, если будет получена вакцина, вызывающая выработку специфических антител к какому-то набору из этих консервативных пептидных участков, и, если эта вакцина будет обладать вируснейтрализующими свойствами, то проблема вирусной изменчивости не будет играть существенной роли. Необходимое для получения вакцин количество таких полипептидов сейчас легко может быть получено методами генной инженерии или химическим синтезом. Кроме того, предполагается экспрессия таких консервативных пептидов в генетически модифицированных растениях с целью получения “съедобных вакцин”. Еще одним перспективным вариантом было бы создание иммуноглобулиновых сывороток на основе моноклональных антител специфичных к таким консервативным пептидным участкам вирусных белков.
Кроме того, было установлено, что консервативные участки белков - это участки нуклеации, т.е. участки, которые играют наиболее важную роль, когда из линейной структуры белковая молекула превращается в глобулу, приобретая необходимую пространственную структуру (фолдинг). Консервативные участки существуют в белках всех без исключения вирусных вариантов. У разных вирусов они разные, но у одного типа вируса они одинаковы. На основе этого факта может быть создан новый класс лекарств, который будет блокировать фолдинг вирусных белков. Это может стать основным путем, который позволит бороться с вирусными инфекциями.
В последнее время большое внимание уделяется разработке новых антимикробных средств, действующих как ингибиторы белок-белковых взаимодействий. Подобно консервативным пептидам, обеспечивающим фолдинг, участки полипептидной цепи различных белков, обеспечивающие их взаимодействие друг с другом (димеризацию, олигомеризацию) при образовании комплексов практически не подвержены мутациям. Поэтому возможно значительное снижение риска появления лекарственной резистентности, обусловленной мутациями такой белковой мишени. Считается, что предпочтительно выбирать в качестве мишеней ферменты, функционирующие в гомо-, гетеро- или олигомерной форме.
Основными молекулярными мишенями для действия имеющихся сейчас анти-ВИЧ препаратов служат вирусоспецифические белки - обратная транскриптаза, и протеаза вируса. Однако аналоги этих ферментов имеются у человека, поэтому препараты, ингибирующие вирусные белки (азидотимидин, d4T и др.), токсичны и для человека. Более перспективной мишенью является фермент интеграза, обеспечивающая интеграцию линейной провирусной ДНК в геном (хромосому) человека. В клетках органов человека такого фермента нет, поэтому препараты ингибирующие интегразу должны быть значительно менее токсичны для человека. В ГНЦ “Вектор” (г.Новосибирск) и Новосибирском институте органической химии СО РАН получены вещества – ингибиторы этого фермента. Проведенные исследования показали, что амиды бетулоновой и глицеризиновой (корень солодки) кислот, а так же дипептид бетулоновой кислоты эффективно ингибируют интегразу ВИЧ-1. В настоящее время проводятся доклинические исследования соединения, наиболее перспективного для клинического применения.
Достаточно сложной представляется и задача создания эффективных препаратов для лечения герпесвирусных инфекций. Это обусловлено тем, что представители этого семейства вирусов имеют большой размер генома, контролирующий синтез более 100 белков, и широкий спектр ферментов, участвующих в метаболизме нуклеиновых кислот, синтезе вирусной ДНК и экспрессии белков. Представленные сейчас на рынке противогерпесные химиопрепараты прямого действия (ацикловир, ганцикловир и др.), подавляющие синтез вирусной ДНК, являются токсичными, к ним может возникать устойчивость и они не способны предотвратить рецидивы. В связи с этим в последнее время проведены широкие исследования по поиску новых препаратов различной природы (антибиотики, пептиды, растительные субстанции, производные фуллерена-60). Было показано, что поиск препарата действующего на одну, определенную мишень, является длительным и неэффективным. Более перспективным представляется создание комплексного препарата, одновременно направленного на несколько мишеней в вирусе.
В связи с этим сотрудниками Научно-иновационного фонда Здоровья (г. Москва) предложена новая концепция многоуровневой блокады проникновения вирусов в клетки. По мнению ученых, анализ молекулярной организации вирусов позволяет идентифицировать их вирионы, как самоорганизующиеся мульти-интер-биополимерные комплексы наноразмерного уровня (-20-500 нм), на котором и реализуются механизмы паразитической интервенции в клетки. Эта величина на несколько порядков больше размеров молекул низкомолекулярных лекарств, что не обеспечивает полноценной защиты клеток организма от вирусов (мутации которых выводят вирусные структуры из-под моноточечных воздействий несоразмерно малых молекул). Поэтому была разработана принципиально новая стратегия противовирусной защиты, (блокады проникновения), основанная на трех фундаментальных принципах: (1) принцип использования вирус-специфических ингибиторов, представляющих собой высокомолекулярные соединения - полиэлектролиты, соизмеримые с белковыми комплексами вирусов; (2) принцип комбинированного воздействия, по крайней мере, на несколько макромолекулярных мишеней, критически существенных в жизненном цикле вирусов; (3) принцип упреждающей терапевтической блокады вирусов до их проникновения в клетки. В рамках этой стратегии впервые был создан ряд новых синтетических и полусинтетических высокомолекулярных соединений (ВМС) сочетающих в себе комплекс вирус-блокирующих химических структур и функций, которые обеспечивают: стимулирование иммунного ответа, подавление абсорбции вирионов на плазматической мембране, (и) блокаду вирусной фузии на клеточных мембранах посредством полимерсвязанных мембранопротекторов каркасного и терпеноидподобного строения, фокусировку концентрации и активности новых лекарств в эпицентрах повышенного риска вирусного вторжения - в нано-размерных "raft''-доменах липидных оболочек вируса и(или) клеточных мембран, и конкурентную имитацию используемых вирусами клеточных рецепторов - путем введения в структуру высокомолекулярных лекарственных препаратов синтетических пептидных имитаторов таких рецепторов (например, ВИЧ-специфичных CCR5/CXCR4). Эти вещества могут найти применение как микробицидные добавки в различные лекарственные формы (вагинальные свечи, кремы, мази, смазки презервативов и т.д.) Экспериментальные испытания (in vitro/vivo) новых систем подтвердили их уникально высокий потенциал противовирусной защиты в отношении широкого спектра вирусов, включая ВИЧ.
Противовирусные препараты — соединения природного или синтетического происхождения, применяющиеся для лечения и профилактики вирусных инфекций. Действие многих из них избирательно направлено на различные стадии развития вирусной инфекции и жизненного цик
Противовирусные препараты — соединения природного или синтетического происхождения, применяющиеся для лечения и профилактики вирусных инфекций. Действие многих из них избирательно направлено на различные стадии развития вирусной инфекции и жизненного цикла вирусов.
В настоящее время известно более 500 вирусов, возбудителей заболеваний человека. Вирусы содержат одно- или двухцепочечную рибонуклеиновую кислоту (РНК) или дезоксирибонуклеиновую кислоту (ДНК), заключенную в белковую оболочку — капсид. У некоторых из них есть и внешняя оболочка из липопротеидов. Многие вирусы содержат ферменты или гены, обеспечивающие репродукцию в клетке-хозяине. В отличие от бактерий у вирусов нет собственного обмена веществ: они используют метаболические пути клетки-хозяина.
РНК-содержащие вирусы или синтезируют матричную РНК (мРНК), или сама РНК выполняет функцию мРНК. На ней синтезируются вирусные белки, в том числе РНК-полимераза, при участии которой образуется мРНК вируса. Транскрипция генома некоторых РНК-содержащих вирусов осуществляется в ядре клетки-хозяина. Под действием обратной транскриптазы ретровирусов на основе вирусной РНК синтезируется комплементарная ей ДНК (провирус), которая встраивается в геном клетки-хозяина. В дальнейшем при транскрипции образуется как клеточная РНК, так и мРНК вируса, на которой синтезируются вирусные белки для сборки новых вирусов. Вирусы и заболевания, которые ими вызываются, отражены в табл. 1.

На стадии заражения вирус адсорбируется на клеточной мембране и проникает в клетку. В этот период применяются препараты, нарушающие этот процесс: растворимые ложные рецепторы, антитела к мембранным рецепторам, ингибиторы слияния вируса с клеточной мембраной.
На следующем этапе начинается внутриклеточный синтез вирусных компонентов. На этом этапе эффективны ингибиторы вирусных ДНК-полимераз, РНК-полимераз, обратной транскриптазы, геликазы, праймазы, интегразы. На трансляцию вирусных белков действуют интерфероны (ИФН), антисмысловые олигонуклеотиды, рибозимы и ингибиторы регуляторных белков. На протеолитическое расщепление воздействуют ингибиторы протазы.
ИФН и ингибиторы структурных белков активно воздействует на сборку вируса.
Заключительный этап репликационного цикла включает выход дочерних вирионов из клетки и гибель инфицированной клетки-хозяина. На этом этапе эффективны ингибиторы нейраминидазы, противовирусные антитела и цитотоксические лимфоциты.
Существуют различные классификации противовирусных средств. В данной статье представлена классификация по воздействию на тот или иной вирус (табл. 2).

Рассмотрим противогриппозные и противогерпетические препараты.
Классификация противовирусных препаратов, разрешенных к применению на территории России.
- руппа противогриппозных препаратов:
– Амантадин;
– Арбидол;
– Осельтамивир;
— Римантадин. - Препараты, действующие на герпесвирусы:
– Алпизарин;
– Ацикловир;
– Бонафтон;
– Валацикловир;
– Ганцикловир;
– Глицирризиновая кислота;
– Идоксуридин;
– Пенцикловир;
– Риодоксол;
– Теброфен;
– Тромантадин;
– Фамцикловир;
– Флореналь. - Антиретровирусные препараты:
– Абакавир;
– Ампренавир;
– Атазанавир;
– Диданозин;
– Залцитабин;
– Зидовудин;
– Индинавира сульфат;
– Ламивудин;
– Нелфинавир;
– Ритонавир;
– Саквинавир;
– Ставудин;
– Фосфазид;
– Эфавиренз. - Другие противовирусные препараты:
– Инозин пранобекс;
– Интерферон альфа;
– Интерферон альфа-2;
– Интерферон альфа-2b;
– Интерферон бета-1а;
– Интерферон бета-1b;
– Йодантипирин;
– Рибавирин;
– Тетраоксо-тетрагидронафталин (Оксолин);
– Тилорон;
– Флакозид.
Арбидол — производное индолкарбоновой кислоты. Механизм действия препарата складывается из подавления репродукции вируса гриппа, влияния на синтез ИФН, повышения количества Т-лимфоцитов и функциональной активности макрофагов, а также антиоксидантного эффекта.
Препарат проникает в неизмененном виде как в незараженные, так и в зараженные клетки и определяется в ядерной и цитоплазматической фракциях. Арбидол ингибирует процесс слияния липидной вирусной оболочки с мембранами эндосом (при рН 7,4), приводящий к высвобождению вирусного генома и началу транскрипции. В отличие от амантадина и римантадина, Арбидол ингибирует освобождение самого нуклеокапсида от наружных белков, нейраминидазы и липидной оболочки. Таким образом, Арбидол действует на ранних стадиях вирусной репродукции.
У препарата отсутствует штаммовая специфичность (в культурах клеток он подавляет репродукцию вируса гриппа А на 80%, вируса гриппа В — на 60% и вируса гриппа С — на 20%, а также воздействует и на вирус птичьего гриппа, однако слабее, чем на репродукцию человеческих штаммов вируса гриппа).
Синтез ИФН нарастает, начиная с приема 1 таблетки до 3 таблеток. Однако дальнейшего увеличения уровня ИФН при приеме Арбидола не наблюдается. Быстрое нарастание синтеза ИФН может оказывать профилактическое действие при приеме препарата до начала заболевания гриппом.
Арбидол оказывает иммуномодулирующее действие, приводя к повышению общего количества Т-лимфоцитов и Т-хелперов. Причем нормализация данных показателей наблюдалась у пациентов с исходно сниженным числом CD3- и CD4-клеток, а у лиц с нормальным функционированием клеточного звена иммунитета практически отсутствовали изменения количества Т-лимфоцитов и Т-хелперов. При этом применение Арбидола не ведет к существенному снижению абсолютного числа Т-супрессорных лимфоцитов — таким образом, стимулирующая активность препарата не связана с угнетением функции супрессорных клеток. Арбидол увеличивает общее число макрофагов с поглощенными бактериями и фагоцитарное число. Предполагается, что активирующими стимулами для фагоцитарных клеток явились цитокины и, в частности, ИФН, продукция которого усиливается под воздействием препарата. Увеличивается также содержание натуральных киллеров — NK-клеток, что позволяет характеризовать препарат как индуктор активности естественных киллеров.
Препарат быстро всасывается из желудочно-кишечного тракта (ЖКТ). Т1/2 составляет 16–21 ч. Экскретируется в неизмененном виде с калом (38,9%) и мочой (0,12%). В течение первых суток выводится 90% введенной дозы.
Лекарственные взаимодействия Арбидола с другими лекарственными препаратами в литературе не описаны.
Практически единственными побочными эффектами препарата являются аллергические реакции. Препарат разрешен к применению с 2-летнего возраста.
Арбидол обладает достаточно широким спектром противовирусного действия и используется для профилактики и лечения гриппа типов А и В, в том числе осложненного бронхитом и пневмонией; острых респираторных заболеваний (ОРВИ); хронического бронхита, пневмонии, рецидивирующей герпетической инфекции; в послеоперационном периоде — для нормализации иммунного статуса и профилактики осложнений.
Амантадин и римантадин — производные адамантана. Оба препарата даже в малых дозах подавляют репродукцию вируса А. Их противовирусная активность обусловлена двумя механизмами.
Во-вторых, они могут действовать и на этапе сборки вируса, по-видимому, за счет изменения процессинга гемагглютинина. Этот механизм возможен у некоторых штаммов вирусов.
Среди диких штаммов устойчивость к препаратам возникает редко, однако от больных, принимающих их, получают устойчивые штаммы. Чувствительность и устойчивость вирусов к амантадину и римантадину перекрестная.
Оба препарата хорошо всасываются при приеме внутрь, имеют большой объем распределения. Большая часть амантадина выводится с мочой в неизмененном виде. Период полувыведения (Т1/2) у молодых людей составляет 12–18 ч, у пожилых возрастает почти вдвое, а при почечной недостаточности увеличивается еще больше. Поэтому дозу препарата необходимо уменьшать даже при незначительном изменении функции почек. Римантадин активно метаболизируется в печени, Т1/2 в среднем составляет 24–36 ч, 60–90% препарата выводится с мочой в виде метаболитов.
При приеме обоих препаратов наиболее часто отмечают незначительные дозозависимые нарушения со стороны ЖКТ (тошнота, снижение аппетита) и центральной нервной системы (ЦНС) (раздражительность, бессонница, нарушение концентрации внимания). При приеме высоких доз амантадина возможно значительное нейротоксическое действие: спутанность сознания, галлюцинации, эпилептические припадки, кома (эти эффекты могут усиливаться при одновременном приеме Н1-блокаторов, М-холиноблокаторов, психотропных средств и этанола). Безопасность применения во время беременности не установлена. Разрешено применение с 7-летнего возраста.
Препараты применяются для профилактики и лечения гриппа А. Их прием во время эпидемий гриппа позволяет избежать инфекции в 70–90% случаев. У лиц с неосложненным гриппом А лечение препаратами в течение 5 дней в возрастных дозировках, начатое на ранней стадии заболевания, на 1–2 сут уменьшает длительность лихорадки и общих симптомов, ускоряет выздоровление и иногда сокращает период выделения вируса.
Осельтамивир является неактивным предшественником, который в организме превращается в активный метаболит — осельтамивира карбоксилат. Он является переходным аналогом сиаловой кислоты и избирательным ингибитором нейраминидазы вирусов гриппа А и В. Кроме того, он подавляет штаммы вируса гриппа А, устойчивые к препаратам — производным адамантана.
Нейраминидаза вируса гриппа отщепляет концевые остатки сиаловых кислот и, таким образом, разрушает рецепторы, находящиеся на поверхности клеток и новых вирусов, т. е. способствует выходу вируса из клетки по окончании репродукции. Активный метаболит осельтамивира вызывает изменения в активном центре нейраминидазы и подавляет ее активность. Происходит агрегация вирусов на поверхности клетки и замедляется их распространение.
Устойчивые штаммы вируса гриппа А обнаруживают у 1–2% больных, принимающих препарат. Устойчивых штаммов вируса гриппа В на сегодняшний день не обнаружено.
При приеме внутрь препарат хорошо всасывается. Прием пищи не влияет на его биодоступность, но снижает риск побочного действия на ЖКТ. Препарат подвергается ферментативному гидролизу в ЖКТ и печени с образованием активного метаболита. Объем распределения препарата приближается к объему жидкости в организме. Т1/2 осельтамивира и его активного метаболита составляет 1–3 и 6–10 ч соответственно. Оба соединения выводятся главным образом почками в неизмененном виде.
При приеме внутрь возможны незначительные неприятные ощущения в животе и тошнота, которые уменьшаются при приеме препарата во время еды. Желудочно-кишечные расстройства обычно проходят через 1–2 сут, даже если больной продолжает прием препарата. Клинически значимых взаимодействий осельтамивира с другими препаратами не выявлено. Препарат применяют у детей старше 1 года.
Осельтамивир применяют для лечения и профилактики гриппа. Профилактический прием осельтамивира в период эпидемий снижает заболеваемость как среди вакцинированных противогриппозной вакциной, так и среди невакцинированных. При лечении гриппа этим препаратом выздоровление наступает на 1–2 сут раньше, а количество бактериальных осложнений снижается на 40–50%.
Прежде чем перейти к обсуждению противогерпетических средств, необходимо вспомнить различные вирусы герпеса и заболевания, вызываемые ими (табл. 4). К сожалению, в арсенале современных противовирусных средств нет препаратов, действующих на все вирусы герпеса одновременно (табл. 5).


Вирус простого герпеса типа 1 вызывает поражение кожи, рта, пищевода и головного мозга, вирус простого герпеса типа 2 — поражение наружных половых органов, прямой кишки, кожи и мозговых оболочек. Первым из допущенных к применению противогерпетических препаратов был видарабин (1977). Однако ввиду высокой токсичности его применяли для лечения заболеваний, вызванных вирусом простого герпеса и Varicella–zostervirus, лишь по жизненным показаниям. С 1982 г. для лечения больных с менее тяжелым течением заболевания стали применять ацикловир.
Ацикловир — ациклический аналог гуанозина, а валацикловир — L-валиновый эфир ацикловира. Ацикловир подавляет синтез вирусной ДНК после фосфорилирования вирусной тимидинкиназой внутри зараженных клеток. Образующийся в клетке ацикловиртрифосфат встраивается в синтезируемую в клетке-хозяине цепь ДНК, что приводит к прекращению роста вирусной цепи ДНК. Молекула ДНК, в состав которой входит ацикловир, связывается с ДНК-полимеразой, необратимо инактивируя ее.
Устойчивость вируса может возникнуть в результате снижения активности вирусной тимидинкиназы и изменения вирусной ДНК-полимеразы. Изменение активности ферментов возникает в результате мутаций.
Биодоступность ацикловира при приеме внутрь составляет всего 10–30% и уменьшается с увеличением дозы. В отличие от ацикловира, биодоступность валацикловира при приеме внутрь достигает 70%. Препарат быстро и почти полностью превращается в ацикловир. Ацикловир проникает во многие биологические жидкости, в том числе в содержимое везикул при ветряной оспе, спинно-мозговую жидкость, накапливается в молоке, околоплодных водах и плаценте. Концентрация его во влагалищном содержимом колеблется в широких пределах. Сывороточная концентрация препарата у матери и новорожденного примерно одинаковы. Через кожу препарат практически не всасывается. Т1/2 ацикловира составляет в среднем у взрослых 2,5 ч, у новорожденных — 4 ч, у больных с почечной недостаточностью может увеличиваться до 20 ч. Препарат практически полностью выводится почками в неизмененном виде. При беременности фармакокинетика препаратов не меняется.
Как правило, ацикловир переносится хорошо. При применении мази на основе полиэтиленгликоля возможно раздражение слизистой половых органов и чувство жжения. При приеме внутрь препарат изредка вызывает головную боль, головокружение, сыпь и диарею. Еще реже отмечаются почечная недостаточность и нейротоксическое действие. Побочные эффекты валацикловира сходны с таковыми у ацикловира — тошнота, диарея, головная боль; высокие дозы могут вызвать спутанность сознания, галлюцинации, поражения почек и — очень редко — тромбоцитопению. При внутривенном введении больших доз ацикловира могут развиться почечная недостаточность и поражения ЦНС.
Фамцикловир сам неактивный, но при первом прохождении через печень быстро превращается в пенцикловир. Пенцикловир — это ациклический аналог гуанозина. Механизм действия препарата сходен с механизмом действия ацикловира. Как и ацикловир, пенцикловир действует главным образом на вирусы простого герпеса и Varicella–zostervirus. Устойчивость к пенциклавиру в клинике встречается редко.
В отличие от пенцикловира, биодоступность которого при приме внутрь составляет лишь 5%, фамцикловир хорошо всасывается. При приеме фамцикловира биодоступность пенцикловира возрастает до 65–77%. Прием пищи совместно с препаратом замедляет всасывание последнего, но в целом биодоступность не снижается. Объем распределения пенцикловира в 2 раза превышает объем жидкости в организме, Т1/21/2 увеличивается до 9,9 ч. Препарат легко удаляется при гемодиализе.
Переносится ацикловир хорошо, но иногда возможно возникновение головной боли, тошноты, диареи, крапивницы, а у пожилых людей — галлюцинаций и спутанности сознания. Препараты для местного применения могут вызвать контактный дерматит и изъязвления.
Безопасность препарата во время беременности, а также взаимодействие его с другими лекарственными средствами не установлена.
Ганцикловир — это ациклический аналог гуанозина. Механизм действия препарата сходен с механизмом действия ацикловира. Активен в отношении всех герпесвирусов, но наиболее эффективен в отношении цитомегаловируса.
Биодоступность ганцикловира при приме внутрь во время еды составляет 6–9% и несколько меньше при приеме натощак. Валганцикловир хорошо всасывается и быстро гидролизуется до ганцикловира, биодоступность которого возрастает до 61%. При приеме валганцикловира во время еды биодоступность ганцикловира повышается еще на 25%. При нормальной функции почек Т1/2 составляет 2–4 ч. Более 90% препарата выводится почками в неизмененном виде. При почечной недостаточности Т1/2 увеличивается до 28–40 ч.
Основной дозалимитирующий побочный эффект ганцикловира — угнетение кроветворения (нейтропения, тромбоцитопения). У 5–15% больных отмечают поражения ЦНС разной степени тяжести (от головной боли до судорог и комы). При внутривенном введении возможны флебиты, азотемия, анемия, сыпи, лихорадка, изменение биохимических показателей печени, тошнота, рвота, эозинофилия.
У лабораторных животных препарат оказывал тератогенное и эмбриотоксическое действие, необратимо нарушал репродуктивную функцию. Цитостатические препараты усиливают побочное действие ганцикловира на костный мозг.
Идоксуридин — йодсодержащий аналог тимидина. Механизм противовирусного действия до конца не изучен. Известно, что фосфорилированные производные препарата встраиваются в вирусную и клеточную ДНК, но ингибируют репликацию только вирусной ДНК. При этом ДНК становится более хрупкой, легко разрушается, при ее транскрипции чаще возникают ошибки. Устойчивые штаммы выделяют от больных герпетическим кератитом, получавших идоксуридин. Препарат разрешен лишь для местного применения. При его использовании возможны боль, зуд, воспаление и отек в области глаз, аллергические реакции.
Успехи антимикробной терапии ХХ столетия привели к почти полному контролю над бактериальными инфекциями. Задачей инфекционистов и фармакологов ХХI века является обеспечение контроля над вирусной инфекцией. Помимо высокой эффективности новые противовирусные препараты должны обладать хорошей переносимостью. В настоящее время разрабатываются новые средства с принципиально новыми механизмами действия. Перспективными могут оказаться средства для подавления патологических иммунных реакций и иммунотерапия моноклональными антителами и вакцинами.
Н. М. Киселева, кандидат медицинских наук, доцент
Л. Г. Кузьменко, доктор медицинских наук, профессор
РГМУ, Москва
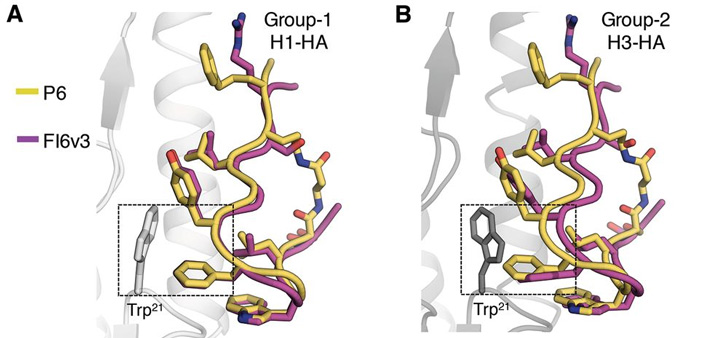
Рис. 1. Сравнение взаимодействия ранее изученного антитела человека FI6v3 и разработанного пептида с частицами вирусов гриппа. Совмещение молекул демонстрирует сходство и различия в том, как антитело (лиловое) и синтезированный пептид (желтый) связываются с гемагглютининовым рецептором вирусов H1 (А) и H3 (B). Оба типа рецепторов, характерные для различных штаммов вируса, связываются с антителом и синтетическим пептидом за счет остатка аминокислоты триптофана (Trp 21 ), положение которого в первичной структуре геммагглютининов Н1 и Н3 одинаково. Рисунок из обсуждаемой статьи в Science
Международная группа исследователей из США, Бельгии и Нидерландов разработала искусственные пептиды, способные нейтрализовать широкий спектр вирусов гриппа, включая штамм H1N1, вызвавший пандемию 2009 года, и штамм высокопатогенного птичьего гриппа H5N1. Эти пептиды могут стать новым оружием в борьбе человечества с гриппом, только сезонные эпидемии которого, по оценкам Всемирной организации здравоохранения, ежегодно приводят к 3–5 миллионам случаев тяжелой болезни и госпитализации и 250 000–500 000 случаев смерти во всем мире.
Существующие в настоящее время способы лекарственной терапии гриппа основаны на воздействии на два белка вируса — канал M2 и нейраминидазу (NA). Оба эти белка важны для нормальной организации жизненного цикла вируса. Белковый канал M2 бывает задействован на ранней и поздней стадиях репликации вируса, а нейраминидаза участвует в высвобождении дочернего поколения вирионов. Но мутации вирусов гриппа приводят (и уже привели) к появлению резистентных штаммов, против которых существующие противовирусные препараты (даже самый эффективный из существующих на настоящий момент — осельтамивир) оказываются малоэффективными. Эти обстоятельства приводят к необходимости разработки новых способов борьбы с вирусами гриппа. Предполагается, что более эффективными могут оказаться стратегии, опирающиеся на применение препаратов, молекулярной мишенью которых будут белки-гемагглютинины (НА) вируса гриппа.
Гемагглютинины — поверхностные белки вируса гриппа, обеспечивающие способность вируса присоединяться к инфицируемой клетке. Возможно, неспособность вируса внедриться в клетку-хозяина снизит вероятность образования устойчивых к противовирусным препаратам мутирующих вирусов: вирусы способны размножаться, только паразитируя в клетках, и не попавший в живую клетку вирус будет лишен возможности воспроизводить себя.
Инфицирование вирусом гриппа начинается с того, что гемагглютинины вируса связываются с поверхностью клетки-хозяина. Вирусный гемагглютинин представляет собой гликопротеид (сложный белок, в котором с цепью, состоящей из аминокислотных остатков, связан углеводный фрагмент). После контакта с клеткой гемагглютинин изменяет свое пространственное строение, фактически внедряясь в клеточную мембрану, что, в свою очередь, обеспечивает проникновение вируса внутрь клетки, где он и начинает свой цикл размножения
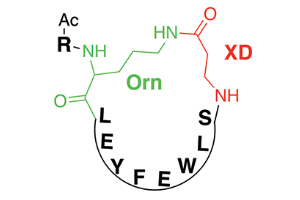
Рис. 2. Общий мотив строения синтезированных циклических пептидов. Цепь пептида состоит из протеиногенных аминокислот — лейцин (L), глутаминовая кислота (Е), тирозин (Y), фенилаланин (F), триптофан (W), серин (S), а также аминокислоты, не входящие в состав белковых молекул: орнитин (диаминовалениановая кислота, Orn) и β-аланин (XD), R — алкильный остаток. Черным цветом обозначена цепь, состоящая из протеиногенных аминокислот, зеленым и красным — участки цепи синтезированного циклического пептида, не характерные для обычных пептидных цепей. С гемагглютинином вируса гриппа циклический пептид связывается преимущественно за счет остатка фенилаланина. Рисунок из обсуждаемой статьи в Science
Ряд спроектированных структур был синтезирован, и после определения их пространственного строения и испытаний на предмет связывания с вирусами исследователи остановились на четырех циклических (обладающих замкнутой цепью, рис. 2) пептидах, каждый из которых обладает свойствами, которые позволяют говорить о его потенциальном применении в качестве блокаторов функций вирусов. Выбранная четверка пептидов демонстрирует прочное связывание с гемагглютининами широкого набора штаммов вирусов гриппа типа А. Синтетические пептиды, как и антитела человека, по образу и подобию которых они были созданы, связываются с остатком аминокислоты триптофана в структуре гемагглютинина вируса. Примечательно, что структура участка, на котором расположен этот триптофановый фрагмент практически не различается для разных штаммов гриппа, поскольку именно этот участок играет ключевую роль в проникновении вируса через оболочку клетки-хозяина.
Основное преимущество разработанных в ходе исследования пептидов по сравнению со ставшими для них образом и подобием антителами заключается в гораздо меньших размерах. Это обеспечивает и дешевизну их получения по сравнению с антителами, и сделает более эффективной усвояемость лекарства при пероральном и инъекционном приеме. Чтобы синтезированные пептиды были устойчивы к действию ферментов, способных ускорять гидролитическое расщепление веществ с аналогичной структурой в крови, в них были введены остатки β-аланина и диаминовалериановой кислоты (орнитина) — аминокислот, не кодируемых в ходе синтеза белковых цепей на рибосоме. Циклическая структура синтезированных пептидов, отсутствие у них характерных для линейных и разветвленных белковых молекул C- и N-конца также способствует устойчивости к гидролизу.
Читайте также: