
Белки как инфекционные агенты
- 13738
- 11,8
- 21
- 4
Путь прионов

Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Биологическая сущность прионов
Рисунок 1. Метафора нейродегенеративного поражения мозга — это губка, в которую превращается нервная ткань в результате массовой гибели нейронов.
И тогда происходит удивительное событие: нормальные молекулы белка, контактируя с патологическими, сами превращаются в них, изменяя свою пространственную структуру (механизм трансформации остаётся загадкой и по сей день) [1]. Таким образом прион, как самый настоящий инфекционный агент, заражает нормальные молекулы, запуская цепную реакцию, разрушительную для клетки.
Некоторые сведения о прионах
Условия возникновения заболеваний
Условия возникновения прионовых болезней уникальны. Они могут формироваться по трём сценариям: как инфекционные, спорадические и наследственные поражения. В последнем варианте главную роль играет генетическая предрасположенность [2].
В последнее десятилетие интерес к этой теме возобновился в связи с возможностью развития диагностики и эффективной терапии [5]. Появилось множество различных объяснений для возрастных нейродегенеративных болезней, — например, окислительная модификация ДНК, липидов и/или белков; соматические мутации; измененный врождённый иммунитет; экзогенные токсины; несоответствия ДНК—РНК; нарушение работы шаперонов; отсутствие одного из аллелей гена [5]. Альтернативным комплексным разъяснением служит то, что различные группы белков могут формировать прионы. Несмотря на то, что небольшое количество прионов может быть удалено посредством путей белковой деградации, их чрезмерное накопление с течением времени позволяет прионам самостоятельно распространяться в организме (рис. 2), что приводит к нарушению деятельности центральной нервной системы [5].

Группы риска прионных заболеваний
Вот кого прионные заболевания могут настичь с наибольшей вероятностью:
- работники пищевой промышленности;
- ветеринары;
- патологоанатомы;
- хирурги;
- пациенты трансплантолога;
- каннибалы;
- лица, в семье которых были замечены синдромы Герстманна—Штрейслера—Шейнклера или фатальной инсомнии.
Лабораторная диагностика и лечение
Диагностика базируется на внутримозговом заражении мышат или хомяков, у которых медленно (до 150 дней) развивается соответствующее заболевание, если пациент был болен [2]. Часто проводится гистологическое исследование головного мозга погибших животных [2].
К сожалению, до настоящего времени еще не разработаны эффективные методы лечения прионовых болезней, хотя попытки предотвратить конформационный переход нормального белка в аномальный производятся. Поэтому самым надёжным способом предупреждения развития инфекционных форм является профилактика [2].
Перспективы
По-видимому, интерес к прионам не угаснет до тех пор, пока предположения на их счёт полностью не подтвердятся и не будут найдены эффективные способы лечения прионных заболеваний. В статье [6] говорится о необходимости современного исследования, которое требует тщательного рассмотрения чужеродных прионов в экстраневрональных тканях.
В качестве модельных объектов авторы использовали мышей: две линии, которые трансгенно экспрессировали овечий прионный белок, и одну линию, которая экспрессировала человеческий прионный белок (рис. 3). Задачей было сравнить эффективность межвидовой передачи инфекции посредством тканей мозга и селезёнки. Внутримозговое заражение чужеродным прионным белком выражалось в отсутствии или небольшом количестве инфекционного агента в мозгах этих мышей. Однако инфекционные чужеродные прионы обнаруживались в селезёнке на более ранних этапах заражения в сравнении с моментом, когда были использованы нейротропные прионы, тем самым определяя, что лимфатическая ткань может быть более пермиссивной к распространению чужеродных прионов по сравнению с мозгом.
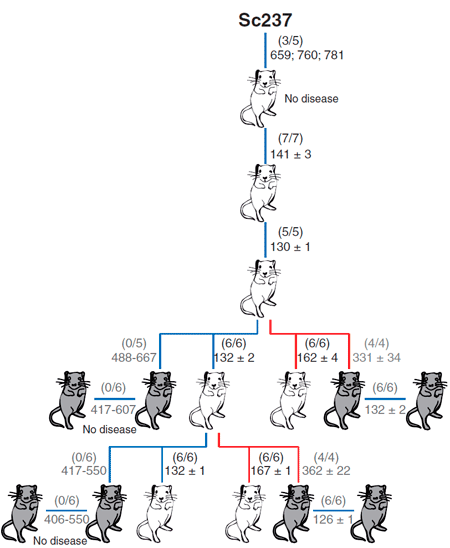
Рисунок 3. Способность приона хомяков Sc237 заражать и передаваться при введении в мозг или селезенку трансгенным мышам, имеющим прионный белок PrP овцы (tg338; белые мыши) или человека (tg7; серые мыши). Число заболевших/инъецированных мышей показано в скобках; ниже приведено среднее время жизни (в днях).
Чем вызвана эта предпочтительная репликация прионов в лимфатических тканях, пока неизвестно. Однако полученные данные показывают, что человек может быть более чувствительным к чужеродным прионам, чем предполагалось ранее на основании присутствия прионов в мозгу, и по этой причине бессимптомный переносчик прионной болезни может быть не распознан. Это ещё раз подтверждает, что такая могущественная биомолекула как прион таит в себе немало загадок, раскрытие которых, возможно, поможет в понимании ряда неразрешимых проблем человечества.
Прион- особая форма инфекционного агента, которая представлена низкомолекулярным белком, имеющим аномальную, по сравнению с белками человека, третичную структуру. Прионы лежат в основе развития медленных инфекций, т.е. инфекций, имеющих длительный период развития и заканчивающихся всегда летально. Главным отличием прионов от других инфекционных агентов является отсутствие у них как такого генетического материала, заключенного в нуклеиновых кислотах (ДНК и РНК). Прионы полностью состоят из белков с низкой молекулярной массой, они не вызывают воспаления и иммунного ответа, а также устойчивы ко многим видам воздействий (высокие температуры, излучение и т.д.). Считается, что прионы могут синтезироваться каждой клеткой организма, но в норме ген, ответственный за воспроизводство прионов репрессирован.
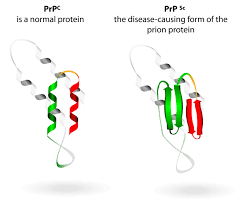
Разница в конформации нормального и патогенного белка
Прионы, соответственно, вызывают прионные заболевания. Прионные заболевания относятся к нейродегенеративным заболеваниям, в процессе развития которых в нервной ткани накапливается аномальной структуры белок. Это происходит вследствие того, что инфекционный прионовый белок PrPsc вызывает мутацию гена, который кодирует нормальный нейрональный прионный белок PrPc, в результате чего клетка начинает синтезировать PrPsc, который отличается от нормального нарушенной третичной структурой. Также PrPsc способен распространяться по типу зомби-вируса из голливудских фильмов, контактируя с нормальным PrPc, он вызывает изменение его конформации и превращение в PrPsc, который способен поражать новые PrPc, в результате чего скорость распространения инфекции по организму растет в геометрической прогрессии. Если клетки инфицируются одиночными инфекционными молекулами, число молекул РrРSс. образующихся в течение суток, достигает 500-1000, в течение года - до полумиллиона. Это неизмеримо меньше по сравнению со скоростью размножения бактерий и вирусов (многие миллионы частиц в течение часов), что объясняет большую длительность инкубационного периода прионовых болезней. Физиологическое значение белка РrРsс прионовых инфекций связывают с реализацией функций синапсов, сохранением клеток Пуркинье, регуляцией внутриклеточного содержания Са2+ в нейронах, поддержанием трофики некоторых их популяций и сохранением резистентности нейронов и астроцитов к повреждающим факторам. Белок PrP — короткоживущий (период полураспада 5-6 ч). В противоположность этому инфекционный прионовый белок PrPSc накапливается в цитоплазменных везикулах, что приводит к последующему нарушению функций синапсов и развитию глубоких неврологических дефектов. Позднее PrP с высвобождается во внеклеточное пространство и откладывается в амилоидных бляшках.
Прионные инфекции имеют три пути возникновения: заражение, по наследству и спонтанное появление. Чаще всего обнаруживается спонтанное появление прионной инфекции. После инфицирования белок попадает в кровь откуда проникает в мозг, где и накапливается, вызывая характерные изменения. Одной из особенностей инфекции является то, что при заражении не происходит каких-либо изменений в периферической крови, в результате чего затруднена диагностика заболевания на ранних этапах его развития. Макроскопически у больного, умершего от прионной инфекции, определяют атрофические процессы в головном мозге. Микроскопически определяется дегенерация и атрофия, а также утрата нервных клеток с заменой их разрастающийся глиальной тканью. Определяются амилоидные бляшки, содержащие прионный белок. Клинически прионные инфекции проявляются по-разному в зависимости от пораженного участка мозга. Приведем в качестве примера некоторые из них.
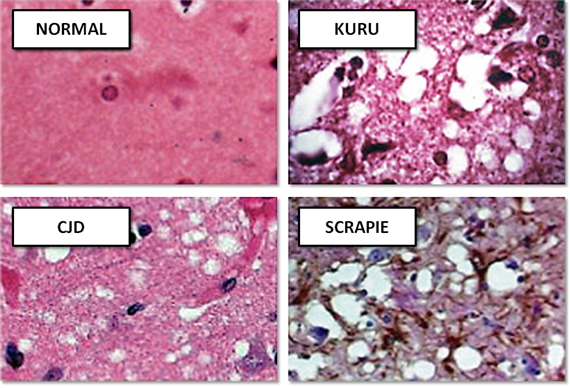
Сравнение гистологических срезов ткани в норме и при различных прионных заболеваниях
Фатальная семейная бессонница. Наследственное заболевание с аутосомно-доминантным типом наследования. При этом заболевании происходит образование амилоидных бляшек в области таламуса. Заболевание развивается медленно и проходит ряд стадий. На первой из них больной жалуется на стойкую бессонницу, при которой не помогают снотворные средства, больной хочет спать, но не может заснуть, позднее возможно присоединение повышенного слюно- и потоотделения, учащение пульса и частоты дыхания, возможно повышение температуры тела. Эта стадия длится около 4х месяцев. Далее к уже имеющийся симптоматике присоединяются панические атаки и галлюцинации. Из-за перенапряжения мозга в связи с отсутствием сна начинаются когнитивные нарушения. Возможно появление непроизвольных движений и подергиваний. Эта стадия длится около 5 месяцев. На следующей стадии сон отсутствует полностью, больной истощен, развивается мышечная слабость, поведение становится неконтролируемым. Это продолжается около 3х месяцев. На заключительной стадии развивается тяжелая деменция, больной перестает ходить, есть. Возможно присоединение вторичной инфекции. Это продолжается около 6 месяцев и неминуемо заканчивается смертью. Заболевание очень редкое, есть данные о наличии этого заболевания у 40 семей итальянского или итало-американского происхождения.

Куру. Ее также называют болезнью каннибалов. Встречается в Новой Гвинеи у аборигенов племени форе. Причинной заболевания является ритуальный каннибализм, во время которого поедается мозг человека, пораженного болезнью. Куру является одной из первых открытых прионных заболеваний. При развитии заболевания постепенно усиливаются головокружение и усталость, затем присоединяются судороги и дрожь. В это время ткани головного мозга постепенно деградируют, превращаясь в губчатую массу (губчатая энцефалопатия). Больные не могут самостоятельно передвигаться, развивается мышечная слабость, на поздних этапах развивается деменция, сходящееся косоглазие и дизартрия. Смерть наступает в течении 2-3 лет. Так как на данный момент ритуальный каннибализм не практикуется, то и болезнь практически исчезла и представляет лишь исторический интерес.

Мальчик больной куру
Болезнь Крейтцфельдта-Якоба- Спорадическое прионное заболевание (т.е. развивающееся без всякой причины), также может возникнуть после употребления зараженного мяса (говядина), возможно заражение при проведении гормональной терапии человеческими гормонами. Ее также называют человеческим вариантом коровьего бешенства, т.к. данный вид прионной болезни вызывает губчатую энцефалопатию у коров. Это заболевание составляет большую часть всех случаев заражений прионными инфекциями (85%). Клинические проявления начинаются с появления раздражительности, рассеянности, головных болей, нарушений сна, изменений поведенческих реакций и т.д. Возможно развитие двигательных нарушений, а в некоторых случаях кратковременной эйфории или депрессии. Позднее развиваются прогрессирующие параличи, эпилептические припадки. Постепенно нарастает деменция, возникают нарушения речи. На поздних стадиях развивается тяжелая деменция, выраженная мышечная атрофия, нарушения глотания, возможно присоединение вторичной инфекции.
К сожалению, в наше время не разработано методов диагностики и лечения прионных инфекций. К счастью, они встречаются очень редко и недостаточно контагиозны, иначе данные инфекции могли бы обернуться настоящей катастрофой с возможным исчезновением всего человеческого рода.
Прионы - это инфекционные агенты белковой природы, которые вызывают Трансмиссивные губкообразные энцефалопатии (ТГЭ) человека и животных [ Prusiner, ea 1991 ]. Уникальная особенность этих заболеваний заключается в том, что они могут возникать не только в результате инфекции, но и спорадически. Далее независимо от происхождения болезни, она передается инфекционным путем. Эти факты, а также чрезвычайная устойчивость прионов к ультрафиолетовому свету впервые позволили предположить, что инфекционной частицей может быть белок. Самой удивительной особенностью прионов является их способность "наследоваться". Прион, попадая в клетку, вызывает изменение конформации своего клеточного аналога, который сам становится прионом. Таким образом, происходит распространение ("наследование") приона при делении клеток. Прионы фактически являются генетическим детерминантом белковой природы.
Основной компонент прионов - аномальная изоформа прионного белка - одного из белков ЦНС. Проникновение прионов в клетку приводит к нарушению конформации синтезируемого клеткой прионного белка , нарушению функции клетки и дальнейшему накоплению прионов. Прионы вызывают такие дегенеративные заболевания ЦНС, как болезнь Крейтцфельдта-Якоба , куру и болезнь Герстмана-Штросслера . Предполагают также участие прионов в передаче человеку губчатой энцефалопатии крупного рогатого скота .
Данные о прионных белках разных организмов представлены в таблице 1 pr .
У млекопитающих прионный белок получил название PrPSc (от болезни scrapie). Он представляет собой особую изоформу нормального клеточного белка PrPC (от cellular), которая отличается плохой растворимостью в детергентах, устойчивостью к действию протеаз, а также склонностью к агрегации [ Тер-Аванесян ea 1999 ]. Из-за сложности работы с прионами млекопитающих строгое доказательство того, что белок PrP является прионом, затруднено, но все имеющиеся факты говорят в пользу этой гипотезы.
В 90-х гг были обнаружены белковые нехромосомно-наследуемые детерминанты у дрожжей, [ Psi+ ] и [ URE3 ] (см. табл. 1 ), для которых практически доказано, что, действительно, носителем наследуемой информации является белок, а вернее его необычное состояние, обладающее свойствами приона. В отличие от других автономных детерминантов дрожжей (плазмид или вирусов), где носителем генетической информации является РНК или ДНК, [Psi+] и [URE3] не удалось связать с какой-либо нуклеиновой кислотой. Они оказлись сходны по ряду принципиальных свйств с прионом млекопитающих.
Эти детерминанты обладают белковой природой, способностью передавать свои свойства нормальной клеточной форме белка, возникать самопроизвольно из клеточной формы, а также обладают устойчивостью к различным агентам, как-то к протеазам, УФ и ионизирующему излучению. Все это не характерно для нормальной конформации этих белков. Надо отметить, что существуют различные штаммы прионов, которые различаются своей конформацией и свойствами. Такие различающиеся штаммы могут находиться одновременно в одной клетке. Существуют "слабые" и "сильные" штаммы. Они отличаются устойчивостью к излечивающим агентам и, вероятно, скоростью распространения прионной конформации [ Тер-Аванесян ea 1999 , Kushnirov ea 2000 ].
Что же происходит, когда такой аномальный, прионный белок попадает в клетку или в систему, где есть нормальный клеточный белок - аналог приона? Структурно прионный белок отличается от клеточного только тем, что он обладает другой конформацией, т.е. измененной вторичной и третичной структурой. Для полной конверсии клеточного белка достаточно следового количества прионного белка (далее такое малое количество материала прионного белка будет называться "семенами"). На сегодняшний день практически установлено, что белок в прионной конформации представляет собой агрегат в виде высокоупорядоченных фибрилл, к концам которых может присоединяться нормальный клеточный растворимый белок, и само это связывание, по-видимому, и является тем фактором, который вызывает изменение конформации. При этом белок, находящийся в агрегированном состоянии, не способен далее осуществлять свою нормальную клеточную функцию, в результате чего и возникает [Psi+] или [URE3] фенотип. На дрожжах in vitro было показано, что при добавлении к лизату клеток семян происходит быстрая конверсия - образование агрегатов из всей клеточной формы этого белка. Если же затем взять семена этого агрегата и поместить их в следующую порцию нормального лизата, то опять произойдет конверсия и образование агрегатов. Эту процедуру можно осуществить неограниченное число раз [ Paushkin ea 1997 ].
Однако, на сегодняшний день ясно, что прионные белки являются необходимым, но недостаточным элементом в феномене прионообразования, также важную роль играют и другие белки. Также, по-видимому, необходим целый комплекс факторов, регулирующих образование прионных агрегатов. Существование прионного феномена можно считать практически доказанным, хотя периодически появляются работы, связывающие прионы с вирусами [ Тер-Аванесян ea 1998 ].



Опрос
Хотели бы вы принять участие в федеральной программе "Земский учитель"?
Да, но только на пять лет по контракту, потом уехала(а) бы обратно в город
Да, я мечтаю жить в селе. Это здоровье, простота и доброта людей, близость к природе, малая наполняемость классов и другие плюсы
Хотел(а) бы, но не подхожу по условиям программы. Необходимо их расширить, в частности, по возрасту участников и их образованию
Всего проголосовало: 138
Текущий номер
О подготовленности учителей и образовании
номер 14, от 7 апреля 2020

У педагогов на карантине выходных нет. Они не сидят без дела в соцсетях, не читают хронику эпидемии, не смотрят фильмы, которые рекламируются для скрашивания досуга. Они заняты освоением платформ и составлением видеоуроков, подбором дистанционных заданий, налаживанием каналов обратной связи. И несмотря на загруженность, они с тревогой думают о будущем. Когда и каким будет первый недистанционный урок? Какими вернутся дети? Сможет ли школа выйти из кризиса без потерь?
"Если человек имеет высшее образование как элемент общей культуры, хочет работать в школе и готов потратить время на подготовку к сдаче экзамена на профстандарт, его вполне можно допустить к преподаванию даже авансом при наличии обязательства, скажем, в течение года пройти соответствующие курсы без отрыва от производства", – считает академик РАО Виктор Болотов. Сложностям профстандарта посвящена его авторская колонка в новом номере.
Российские вузы вот уже несколько недель находятся на дистанционном обучении. Что думают о новом формате студенты? Страдает ли качество обучения при удаленной работе? Появилось ли больше времени для личной жизни и дополнительной подготовки? Читайте мнения ребят из разных регионов страны в рубрике "Студсовет"!
В самоизоляции у многих появилось появилось больше времени для чтения. Чтобы перевести внимание, прикованное к пандемии, в более конструктивное русло, представляем авторский обзор книг об эпидемиях, написанных в разное время. Невозможно не заметить, насколько обстоятельства и реалии эпидемий прошлых эпох перекликаются с днем сегодняшним.
Наши приложения
История
1933 год. Из Германии в Исландию завезены овцы для развития каракулеводства. Период адаптации к новым условиям среды всегда связан с повышенной заболеваемостью. Однако скрейпи овец (медленно развивающееся инфекционное заболевание) было выделено отдельно, так как имело долгий, никак не проявляющий себя инкубационный период, сверхмедленное прогрессирующее течение, странное поражение органов (мозг, селезенка) и неотвратимую смерть. Так, в 1954 году Б. Сигурдсон научными исследованиями скрейпи закрепил за собой имя первооткрывателя прионных болезней.
Три года спустя у папуасов Новой Гвинеи обнаружено заболевание куру, связанное с поеданием сырого мозга умерших родственников. Ритуальный каннибализм приводил к неспешно проявляющимся симптомам идентичной скрейпи болезни. К.Гайдушек установил инфекционный путь передачи куру (Нобелевская премия 1976 года).
1992-2000 годы. Достигла максимума эпизоотия среди крупного рогатого скота Великобритании. Мирно пасущиеся животные вдруг начинали носиться по полю, кидаться друг на друга, дико мычать, а затем умирали. Коровье бешенство оказалось той же болезнью, что и известная прежде лишь у овец скрейпи. Выяснилось, что она заразна и для людей - в Европе погибло около 150 человек, заболело 3,8 млн голов крупного рогатого скота. Американец С.Прузинер, открывший возбудитель недуга - патогенные белки-прионы, был удостоен Нобелевской премии 1997 года. Эпизоотию удалось погасить, но лечить это заболевание ни у людей, ни у животных медицина пока не в состоянии. Этот случай привел к созданию жесткой системы контроля мяса. Заодно был обнаружен человеческий аналог коровьего бешенства - болезнь Кройтцфельдта - Якоба, до того неизвестная, поскольку встречается по статистике один случай на миллион. По симптомам она похожа на другие психиатрические недуги (нарушение координации движений, слабоумие).
Биологическое оружие
XXI века?
На сегодня известно, что нормальный прионный белок PrP обнаруживается в клетках цнс, лимфатических тканях. Его патогенная модификация совпадает по аминокислотной последовательности с нормальной, но имеет другую пространственную структуру: при переходе в патогенное состояние часть -спиралей превращается в листоподобные -складчатые слои, образуя множество водородных связей, стабилизирующих инфекционную пространственную структуру. Клеточная функция нормальной формы PrP пока неясна, патогенная форма вызывает заболевания. Инфекционный PrP не только устойчив к деградации (УФ, нагревание, проникающая радиация, протеазы), но также может перестраивать нормальную форму, превращая ее в подобие себя по цепной реакции. Со временем прионы с нарушенной конформацией формируют фибриллы в нервных клетках, приводя к гибели нейронов и в конце концов к смерти особи.
Патогенные формы прионов могут вызывать ряд тяжелых заболеваний: болезнь Кройтифельда - Якоба, семейная фатальная бессонница у человека и губчатая энцефалопатия у крупного рогатого скота, норок, лосей и оленей, кошачьих, экзотических животных, скрейпи овец и коз, которые на данный момент неизлечимы. Практически ничто не способно инактивировать неправильный белок: ни высокие температуры, ни повышенное давление, ни даже обугливание не снижают степень его патогенности. На него не влияют и протеолитические ферменты кишечника. Получается, что животное, употребившее такой белок в пищу, может заразиться само и заразить других особей через экскременты или прах. Напрашивается предположение, что при таком положении дел прионы могут стать мощнейшим биологическим оружием ХХI века.
Доказана внутривидовая передача прионных заболеваний: коровье бешенство при кормлении скота костной мукой, куру при ритуальном каннибализме. Однако иногда инфекция передается межвидово, например, от животных к человеку, хотя при этом болезнь развивается не столь эффективно, реже поражая мозг. Но в любом случае болезнь возникает спорадически, и при ее развитии амилоидные структуры в мозговой ткани разрастаются как губка (отсюда и название - губчатая энцефалопатия). Этот процесс характерен и для недугов пожилого возраста - Альцгеймера и Паркинсона - с печально известными симптомами в виде патологического ухудшения памяти и утраты способности к обучению, поэтому, разгадав загадки прионов, человечество может надеяться если не победить, так отсрочить старение (по И.Мечникову срок человеческой жизни составляет 140 лет).
Загадочный механизм
Белки физиологически существуют в виде пространственных структур, и их конформация слишком важна и сложна, чтобы изменяться случайным образом. Биофизик В.Астбэри когда-то предположил, что нефункциональные фибриллы образуются в результате денатурации (необратимого изменения природной структуры белковой глобулы) под стрессовым воздействием свойств среды. Сейчас известно, что внутриклеточные природные процессы - окисление, фосфорилирование, гликолизирование - могут приводить к изменению термодинамических параметров белков, высвобождая энергию, за счет которой возможен рефолдинг (пересворачивание) белка в неправильную структуру.
К сожалению, в настоящее время механизм, обеспечивающий спорадическое возникновение заболевания, остается загадкой. Пока что человечеству известны наследственные варианты заболевания благодаря открытию гена, кодирующего аминокислотную последовательность PrP. В этом случае происходит мутационное изменение первичной структуры белка, стимулирующее его превращение в прион. Наконец, существует инфекционная форма развития заболевания при проникновении патогенной формы приона в организм.
Светлана ХОРОНЕНКОВА, кандидат химических наук
Как и пациенты с прионным заболеванием, больные Альцгеймером страдают от смертельной прогрессирующей формы деменции. Растет доказательство того, что агрегаты амилоида-β (Aβ) могут быть переданы аналогично прионам, по крайней мере в экстремальных экспериментальных условиях. Однако, в отличие от мышей, инфицированных прионами прионного белка (PrP), инокулированные Aβ не умирают. Таким образом, передача Aβ и PrP заметно отличается от неврологических эффектов, которые они вызывают у своих хозяев, причем разница не меньше, чем вопрос жизни и смерти. Это не просто академический нюанс, это различие между Aβ и PrP вызывает решающие вопросы того, что именно контролирует прионную токсичность и как прионная токсичность относится к инфекционности прионов.
В 1982 году Стэнли Прусинер ввел прионы в качестве самовоспроизводящихся форм прионного белка, которые накапливаются при некоторых трансмиссивных заболеваниях центральной нервной системы, таких как скрепи и болезнь Крейтцфельдта-Якоба1. Хотя прионы представляют собой новые инфекционные агенты, лишенные нуклеиновых кислот, кодируемых патогенами , их открытие опиралось на вековую парадигму, формализованную в постулаты Роберта Коха, для выявления патологических микробных агентов. Ключевая концепция в постулатах Коха заключается в том, что микроб, ответственный за данное заболевание, должен вызывать то же заболевание при инокуляции в восприимчивый хозяин. При прионном заболевании страдающий человек страдает от прогрессирующего ухудшения неврологической функции, которое завершается, неизбежно, в смерти. Путем систематического просеивания мозговых экстрактов из зараженных scrapie хомяков, Прусинер обнаружил, что самые смертоносные инокуляции содержат фибриллярные агрегаты протеолитического фрагмента прионного белка PrP27-30. Теперь мы знаем, что этот фрагмент получен из изоформы scrapie прионного белка PrPSc, агрегированного, альтернативно сложенного конформера клеточного прионного белка, PrPC.2
В 2000 году Лэри Уокер впервые продемонстрировала, что внутримозговые прививки мозговых экстрактов из мозговой ткани, содержащей амилоидный бляшко от пациентов с болезнью Альцгеймера, ускоряют осаждение амилоидных бляшек и β-амилоидоз у трансгенных мышей, экспрессирующих белки человека Aβ3. Ускорение β-амилоидоза инокулятами, содержащими Фибриллы Aβ, которые образуют амилоидные бляшки, были воспроизведены, по меньшей мере, в четырех других лабораториях с использованием инокулятов у людей, нескольких линий формирующих бляшку трансгенных мышей и, в последнее время, фибриллярных синтетических агрегатов Aβ и синтетических димеров Aβ.4-7
Прионы выполняют вышеуказанное определение, поскольку они были первоначально обнаружены как истинные инфекционные агенты с использованием микробиологических методов. Однако многие другие белки могут объединяться в геометрически расположенные структуры, которые могут вносить in vitro и in vivo-компартменты, содержащие исходный белок в мономерном растворимом состоянии.
К счастью, нет никаких признаков того, что такие процессы существуют для прионоидов Aβ. Тем не менее, демонстрация межличностной прозрачности будет способствовать повышению статуса таких агентов для добросовестных прионов, что, по-видимому, очень вероятно в случае амилоида AA.13.
До эпохи молекулярной генетики нейродегенеративные заболевания пожилых людей определялись типами несвязанных белков, которые накапливаются как нерастворимые отложения в мозге. Отложения состояли из высокоупорядоченных стеков β-листов, что является физическим определением амилоида. Каждый тип амилоида находится в характерном ядерном, цитоплазматическом или внеклеточном отделе. С появлением молекулярной генетики было обнаружено причинные гены, связанные со многими из этих нейродегенеративных заболеваний, и в удивительно большом числе случаев данный ген кодировал сам белок, содержащий амилоидные фибриллы, которые характеризовали невропатологию в этом заболевании.
Эти заболевания стали известны как расстройства, связанные с расстройством белков, поскольку каузальные гены кодировали несогласованные белки, включающие амилоидные поражения. В каждом случае несогласованные белки приобретают вторичную структуру-β-листы, которая отсутствует в нормальных условиях. Сопротивление белка в нейродегенеративных заболеваниях относится к условиям, в которых родительские белки берут новую вторичную структуру β-листа. Приобретение новой вторичной структуры отличает расстройства, связанные с расстройством белков, от других заболеваний, вызванных мутациями, которые изменяют конформацию белка, например, серповидноклеточную анемию.
За исключением PrPSc, нет экспериментальных доказательств того, что прионы или прионеиды в нейродегенеративных заболеваниях являются патогенными белками (звездообразными (*) белками], вызывающими неврологическое ухудшение, которое разрушает пациентов. Считается, что столетие нейрофибриллярные клубочки — внутриклеточные амилоидные включения, которые образуются, когда tau принимает новую структуру β-листа, — индуцируют гибель нейронов и ухудшают познание. Однако в 2005 году эта столетняя гипотеза была опровергнута, когда было показано, что уменьшение растворимого тау в модели нейродегенеративной мыши с нейрофибриллярными путаницами привело к прекращению потери нейронов и улучшению функции памяти, несмотря на поразительное наблюдение, что нейрофибриллярные клубочки сохраняли накопление, напоминающее прионеиды.14 Отрицательный случай для β-амилоидных бляшек, содержащих * белки, связан с несколькими линиями доказательств, включая отказ пациентов с болезнью Альцгеймера улучшить после иммунотерапии Aβ, которые, тем не менее, успешно удаляли амилоидные бляшки, 15 и способность иммунотерапии для устранения дефицита у мышей без изменения нагрузки на бляшку.16,17 По сравнению с белками, включающими амилоидные поражения, прионы и прионеиды, относительно мало известно о белках и их механизмах действия.
Безусловно, наиболее понятным среди этих расстройств является семейная атаксия типа 1. Атаксин-1 вызывает семейную атаксию типа 1, когда посторонние последовательности тринуклеотидных повторов расширяют существующий полиглутаминовый тракт с образованием PolyQ / ataxin-1.18. Пациенты и мыши, экспрессирующие PolyQ / ataxin-1, развивают ядерные включений и прогрессирующей атаксии. Агрегаты PolyQ являются прионоидами, поскольку применение агрегатов PolyQ к культивируемым нейронам зарождает образование внутриклеточного полиомиелита амилоида.19 У мышей, несущих разреженный расширенный тракт PolyQ, тяжесть неврологических аномалий и нейродегенераций обратно пропорциональна числу включений, 20 и отмена включений посредством мутации лигида ubiquitin, которая способствует агрегации несогласованных белков, ускоряет заболевание.21 Эти исследования показывают, что элиминация включений не будет препятствовать атаксии в семейной атаксии типа 1.
Вывод о том, что удаление включений не вылечило семейную атаксию типа 1, побудило Гарри Орра и Худу Зогби искать патогенную форму PolyQ / ataxin-1, вызывая неврологические аномалии в расстройстве. Обнаружение механизма, с помощью которого PolyQ / ataxin-1 повреждает нейроны, возникло из понимания нормальной физиологической роли атаксина-1, являющегося ядерным белком. Патогенная форма PolyQ / ataxin-1 не является неправильной формой атаксина-1; он не содержит новых вторичных структур, нет β-листов, которые обычно не присутствуют в мозге. Его патологические эффекты возникают из-за изменений в его аффинности связывания с его нормальными ядерными партнерами, транскрипционного регулятора Capicua и регулятора RNA-сплайсинга RMB17,22, что приводит к изменениям транскриптома, которые, по-видимому, влияют на нейронную функцию и жизнеспособность. Таким образом, патогенная форма PolyQ / ataxin-1 не является прионоидом; он не является ни ложным белком, ни растворимым агрегатом исходного белка. Это может оказаться глубоко важным уроком для всей области исследований нейродегенеративных заболеваний.
Оба пациента с прионным заболеванием и болезнью Альцгеймера страдают от смертельных прогрессирующих форм деменции. Однако, в то время как мыши, зараженные PrP-прионами, умирают, у тех, кто инокулируется с прионами Aβ, нет. Только две гипотезы могут объяснить резкий контраст между коэффициентами летальности, вызванными привитиями PrP и Aβ у мышей.
Агрегаты являются патогенными, но разные агрегаты оказывают влияние на различные клеточные пути. Например, патогенный путь для агрегатов Аβ у людей, отличный от таковых прионов PrP, может отсутствовать у мышей.
Агрегаты не всегда являются патогенными; скорее, варианты исходных белков (* белков) вызывают клеточную дисфункцию, которая приводит к неврологическому заболеванию (рис.1). Эти патогенные варианты не обязательно должны быть неправильными или агрегированными формами исходных белков.
Недавние достижения в нашем понимании нейротоксичности PrP и Aβ благоприятствуют гипотезе 2, как обсуждается ниже.
При прионном заболевании катастрофическая дисфункция мозга связана с глобальным снижением производства белка, вызванным дисрегуляцией eIF2a, фактором инициации трансляции млекопитающих23. Это увлекательное открытие, по-видимому, является механизмом, с помощью которого прионы PrP в конечном итоге вызывают нейротоксичность.
Однако eIF2a локализуется внутри цитозоля, тогда как инфекционные прионы являются внеклеточными. Таким образом, нам все еще остается интересно, как прионы, содержащие патологически агрегированный PrPSc, могут оказывать действие, происходящее из внеклеточной среды, сгибание белков дерьма в эндоплазматическом ретикулуме, индуцировать неожиданно энергичный развернутый белковый ответ и в конечном итоге гасить цитозольный перенос белков. Трудно не заключить, что подавление eIF2a, вероятно, представляет собой нисходящий эффектор патогенного каскада, который инициируется молекулярно и топологически отдаленными событиями.
Рисунок 2. Клеточный прионный белок абсолютно необходим для токсичности инфекционных прионов (А) 39, что подразумевает, что PrPSc оказывает нейротоксичность путем стыковки с PrPC (B). Эта токсичность также может быть вызвана вариантами PrP, встречающимися естественным образом, такими как PrP, несущими сверхматериальные октапептидные повторы (C), или экспериментально сконструированные токсичные варианты, такие как версии PrP, содержащие делеции шарнирной области (D). Недавно было обнаружено, что прионная инфекция приводит к цепочке событий, которая в конечном счете утоляет перенос белка 23, но остается неизвестным, использует ли токсичность, вызванную мутантами PrP (Панели C и D), тот же путь.
Наше понимание нейротоксичности Aβ отстает от нейротоксичности PrP, потому что животные модели, которые пересказывают все аспекты болезни Альцгеймера, которые необходимы для анализа человеческой значимости патологического Aβ, не существуют. У людей патологическая трансформация Aβ инициирует процесс, который включает накопление амилоидных бляшек и часто приводит к фатальному нейродегенеративному состоянию. У мышей образование патологического Аβ может индуцировать осаждение амилоидных бляшек, что отражает присутствие прионоидов Aβ. Удивительно, что накопление прионоидов Aβ у подавляющего большинства мышей не приводит к открытой нейродегенерации, за исключением непосредственной близости от амилоидных бляшек.28-30 У мышей, лишенных синтазы оксида азота 2, накопление априонидов Aβ связано с нейронов в CA3, но не подполе CA1 hippocampal 31, но у людей CA3 сохраняется, а CA1 — нет, 32 ставит под сомнение значимость этой формы смерти нейронов. Независимо от того, является ли смерть нейронов в этой модели из-за априонидов Aβ, также неизвестно. Фатальность, когда она присутствует, является зависимой от деформации и может возникать в отсутствие образования амилоидов, что указывает на существование нейротоксического вида, который не обязательно может быть сопоставим с самораспространяющимися видами.33 Биохимическая идентичность и клеточные эффекты этого форма Aβ остаются неизвестными.
Два типа когнитивной дисфункции развиваются у мышей с образованием Aβ. Один тип встречается у мышей с агрессивным отложением амилоидов — чрезмерное количество прионоидов Аβ, в которых накопленные амилоидные бляшки и их окружающая цитопатология действуют как гигантское космическое поражение в мозге.28 Никакие нейроны не остаются в сердцевинах бляшек и извилистые , дистрофические нейриты и дендриты, частично оголенные шипами, находятся в 50-микронном гало, окружающем ядра (рассмотренные Ashe и Zahs34). Поэтому неудивительно, что за определенным порогом познание изменяется обратно пропорционально нагрузке на бляшку. Однако плотность амилоидных бляшек, необходимых для получения этого эффекта, редко достигается при болезни Альцгеймера, что может объяснить, почему связь между бляшками и потерей или познаниями в нейронах в лучшем случае незначительна.35,36 Чрезмерное накопление прионоидов Aβ у мышей может привести к в когнитивных дефицитах, которые связаны с пространственно-занимающими эффектами амилоидных бляшек, но это отличается от механизма, с помощью которого большая часть нейрональной дисфункции и нейродегенерации происходит у людей с β-амилоидозом или болезнью Альцгеймера.
Другой тип когнитивной дисфункции происходит независимо от бляшек или потери нейронов и, по-видимому, обусловлен нефибриллярной сборкой Aβ 56 см-1 Aβ, обозначенной Aβ * 56.37 Aβ * 56, предполагаемым додекамером, который, скорее всего, образован кластеризацией четырех Aβ-тримеров , Изоляты фибриллярного Aβ в головном мозге не содержат Aβ * 56 (Liu P и Ashe K, неопубликованные данные). Маловероятно, что Aβ * 56 приобретает новую структуру β-листа, поскольку композиционная единица Aβ-тримеры присутствует даже у молодых мышей.37 Отсутствие новой структуры β-листа в Aβ * 56 утверждает, что она является прионеодом, хотя формальная демонстрация потребует экспериментов по передаче. Aβ * 56 не вызывает явной нейродегенерации и все же изменяет познание, нарушая долговременную синаптическую пластичность еще неизвестным механизмом.38 У людей Aβ * 56 можно измерить в CSF, где он увеличивается с нормальным старением и коррелирует умеренно сильно с белком, связывающим микротрубочки тау (М. Ханкоко, М. Грант, А. Валлин, К. Бленноу и К. Эше, неопубликованные данные). Тау высвобождается в CSF, в процессе, который остается неясным, когда нейроны неисправны. CSF tau слабо коррелирует или вообще отсутствует с CSF Aβ1-42, который отражает β-амилоидоз и априониды Aβ.
Хотя было заманчиво постулировать, что Aβ * 56 вызывает последовательность событий, которая приводит к переходу от бессимптомного старения к умеренным когнитивным нарушениям или болезни Альцгеймера, что совпадает с началом открытой нейродегенерации и потерей нейрона, это предсказание не подтвердилось в большое продольное исследование (Handoko M, Grant M, Petersen R и Ashe K, неопубликованные данные). Таким образом, Aβ * 56 нарушает функцию нейронов у мышей и связывается с нейронной неисправностью у людей, но недостаточно для индукции явной нейродегенерации у обоих видов.
В отсутствие животных моделей, содержащих исключительно мутаций, связанных с болезнью Альцгеймера, которые проявляют полный спектр заболеваний, начиная с тонкой дисфункции нейронов и заканчивая фатальной когнитивной опустошением, вопрос о том, требует ли асимптоматический β-амилоидоз Aβ * 56 в полной мере Продуманная болезнь Альцгеймера не может быть рассмотрена экспериментально. Возможно, что одна или несколько неприноидных форм Aβ запускают нейронную дисфункцию и нейродегенерацию при болезни Альцгеймера. Обнаружение этих патогенных форм будет зависеть от создания высококачественных модельных систем болезни Альцгеймера.
При доброкачественных прионных заболеваниях очень большой объем данных связывает агрегированную форму PrP, PrPSc, как с инфекцией прионов, так и прионной нейротоксичностью. Тем не менее, неинфекционные, но нейротоксичные варианты PrP встречаются естественным образом, и более такие варианты были построены экспериментально, что указывает на то, что фенотипическое выражение, типичное для прионных заболеваний, может быть вызвано событиями, происходящими после прионной инфекции. Существует мало свидетельств того, что мыши или люди связывают неврологические эффекты Aβ с зародышеобразующими формами этого белка, в то время как новые данные указывают на конкретную не зародышевую форму Aβ, Aβ * 56, которая вызывает некоторые неврологические признаки болезни. Однако Aβ * 56 не является достаточным для индуцирования неумолимого неврологического ухудшения, которое характеризует болезнь Альцгеймера, указывая на то, что другие критические факторы или формы Aβ работают в сотрудничестве с Aβ * 56 для разрушения мозга. Лечение приона и болезни Альцгеймера будет зависеть от более глубокого понимания патогенных форм PrP и Aβ, которые вызывают дисфункцию мозга, лежащую в основе этих смертельных заболеваний.
Возможные конфликты интересов выявлены не были.
Читайте также: