
Боль в мышцах при миопатии
Воспалительная миопатия — воспалительный процесс, возникающий преимущественно в скелетных мышцах и приводящий к их дегенеративным изменениям. Воспалительная миопатия характеризуется мышечной слабостью, болями в мышцах, резким снижением объема активных движений, развитием контрактур, уплотнением и отечностью мышц. Из методов диагностики воспалительной миопатии наиболее информативно определение уровня КФК и миоглобина, электромиография и биопсия мышц. Лечение воспалительной миопатии осуществляется высокими дозами глюкокортикостероидов с последующей поддерживающей терапией. Однако достаточно часто встречаются резистентные к кортикостероидам формы заболевания.
МКБ-10
- Причины возникновения воспалительной миопатии
- Симптомы воспалительной миопатии
- Диагностика воспалительной миопатии
- Лечение воспалительной миопатии
- Цены на лечение
Общие сведения
Воспалительные миопатии представляют собой целую группу заболеваний, в которую входят как системные поражение мышечной ткани, так и локальные воспалительные процессы в отдельных мышцах. Воспалительная миопатия системного характера представлена дерматомиозитом, полимиозитом, эозинофильным миозитом, миозитом с включениями, ревматической полимиалгией и миопатиями при системных заболеваниях. Примером локальной воспалительной миопатии может быть миозит глазных мышц, псевдотромбофлебит мышц голени и др. Кроме того, воспалительная миопатия может наблюдаться при некоторых инфекционных заболеваниях.
Возраст пациентов, наиболее подверженных заболеваемости, различается в зависимости от вида воспалительной миопатии. Так, полимиозит обычно возникает у лиц старше 30 лет. Дерматомиозит наблюдается как у взрослых, так и в детском возрасте. А миозит с включениями чаще встречается после 40 лет и является наиболее распространенной формой воспалительной миопатии в пожилом возрасте.
Причины возникновения воспалительной миопатии
В некоторых случаях воспалительная миопатия напрямую связана с инфекционным процессом. Агентами, вызывающими инфекционно-воспалительно поражение мышц, могут быть вирусы (краснуха, грипп, ВИЧ-инфекция, энтеровирусы и пр.), бактерии (чаще стрептококковые инфекции), паразитарные инвазии (цистицеркоз, токсоплазмоз, трихинеллез). При этом заболевание классифицируется как инфекционная воспалительная миопатия.
В тех случаях, когда прямая связь миопатии с инфекцией не прослеживается, говорят об идиопатической миопатии. К ней относятся: миозит с включениями, полимиозит, дерматомиозит, миопатии при системных заболеваниях (системной красной волчанке, склеродермии, синдроме Шегрена, системных васкулитах, ревматоидном артрите). Наиболее распространенным как в неврологии, так и в ревматологии является мнение, что идиопатическая воспалительная миопатия имеет аутоиммунный механизм развития. Однако до сих пор не выделен антиген, который запускает лежащую в ее основе аутоиммунную реакцию. В отдельных случаях воспалительная миопатия имеет семейный характер, свидетельствующий о ее генетическом происхождении.
Симптомы воспалительной миопатии
Воспалительная миопатия клинически проявляется болями в мышцах, прогрессирующей мышечной слабостью и тугоподвижностью в пораженных отделах конечностей. Болевой синдром (миалгия) возникает не только в период двигательной активности, но и при прощупывании мышц, а иногда и в состоянии полного покоя. Мышечная слабость, как правило, начинает проявляться затруднением при удерживании предметов в руках. По мере ее прогрессирования для пациента становится невозможным поднять руку или ногу, сесть, встать. Воспалительная миопатия распространенного характера часто приводит к значительному сокращению объема активных движений, которые может выполнять больной, а иногда и к полной обездвиженности.
Воспалительная миопатия протекает с отеком и уплотнением пораженных мышц. В результате воспаления в них происходят атрофические изменения, развивается миофиброз — замещение мышечных волокон на соединительнотканные, возможен кальциноз. Образующиеся мышечные контрактуры приводят к тугоподвижности суставов. Зачастую воспалительная миопатия имеет длительное ремиттирующее течение. В некоторых случаях, чаще у детей, наблюдается полное выздоровление после перенесенного острого приступа воспалительной миопатии.
Наиболее тяжело воспалительная миопатия протекает при вовлечении в воспалительный процесс мышц гортани и дыхательной мускулатуры, что ведет к развитию миопатического пареза гортани и дыхательной недостаточности. Слабость дыхательных мышц приводит к снижению легочной вентиляции и возникновению застойной пневмонии. Слабость мышц глотки является причиной дисфагии (расстройств глотания), при которой возможно попадание пищи в дыхательные пути и возникновение аспирационной пневмонии. Если воспалительная миопатия распространяется на глазодвигательные мышцы, то возникает косоглазие и опущение верхнего века. Поражение мимических мышц приводит к маскообразному выражению лица. Системная воспалительная миопатия, например, дерматомиозит и полимиозит, может сопровождаться поражением сердечной мышцы с клиникой миокардита, кардиомиопатии и сердечной недостаточности.
Полимиозит проявляется типичным для воспалительной миопатии мышечным синдромом, который захватывает мышцы проксимальных отделов конечностей, в первую очередь плечевого и тазового пояса. В большинстве случаев наблюдается также поражение внутренних органов: ЖКТ (диарея, запор, кишечная непроходимость, язва желудка с перфорацией или желудочно-кишечным кровотечением), сердца и сосудов (артериальная гипотония, аритмия, кардиомиопатия и др), легких (очаговая или долевая пневмония). В 15% случаев эта воспалительная миопатия сопровождается суставным синдромом в виде артритов суставов кисти, реже — других суставов конечностей.
Дерматомиозит имеет сходную с полимиозитом клиническую картину. Отличительной чертой является наличие кожных проявлений: эритематозных пятен, участков гипо- и гиперпигментации, очагов гиперкератоза. Возможно поражение слизистых оболочек с развитием стоматита, гингивита, конъюнктивита.
Миозит с включениями характеризуется ранним поражением мускулатуры дистальных отделов рук: сгибателей пальцев и мышц предплечья. В ногах воспалительная миопатия распространяется как на дистальные, так и на проксимальные мышечные группы. Типично поражение разгибателей стопы и четырехглавой мышцы.
Диагностика воспалительной миопатии
К диагностике воспалительной миопатии помимо неврологов могут привлекаться ревматологи, кардиологи, пульмонологи, дерматологи, иммунологи и др. специалисты. К сожалению, на сегодняшний день воспалительная миопатия не имеет четких диагностических критериев. Определенное значение имеет обнаружение в биохимическом анализе крови повышения уровня креатинфосфокиназы (КФК), а также лактатдегидрогеназы и альдолазы, что свидетельствует о повреждении мышечной ткани. Зачастую воспалительная миопатия сопровождается повышением миоглобина. Однако уровень этих показателей не коррелирует с тяжестью клинических проявлений. По этой причине единственным достоверным критерием прогрессирования миопатии могут быть лишь результате исследования мышечной силы (эргометрии) в динамике. Следует также отметить, что повышение КФК и миоглобина наблюдается далеко не при всех видах воспалительной миопатии. Например, при миозите с включениями уровень КФК и миоглобина часто находится в пределах нормы.
Важное диагностическое значение имеют данные электромиографии. При дерматомиозите и полимиозите наблюдается снижение продолжительности и амплитуды потенциалов двигательных единиц, повышенная электровозбудимость. При миозите с включениями наряду с типичными для миопатии низкоамплитудными и кратковременными потенциалами отмечаются высокоамплитудные и длительные потенциалы, характеризующие нейрогенное поражение.
Уточнить характер поражения мышц и определить его распространенность позволяет биопсия мышц. Морфологическое исследование биоптата выявляет наличие воспалительных инфильтратов, атрофические изменения и некроз мышечных волокон, их замещение соединительной тканью.
Диагностика сопутствующих поражений внутренних органов осуществляется при помощи рентгенографии легких, ЭКГ, Эхо-КГ, гастроскопии, УЗИ органов брюшной полости.
Лечение воспалительной миопатии
Общепринятым методом лечения воспалительной миопатии является терапия глюкокортикостероидами. Суточная доза преднизолона составляет 80-100 мг. При достижении клинического эффекта лечения в виде увеличения мышечной силы производят постепенное снижение суточной дозы до поддерживающего уровня — 15 мг/сут. В тяжелых случаях воспалительная миопатия лечится пульс-терапией метилпреднизолоном. Сложности применения длительного лечения глюкокортикоидами состоит в их побочных эффектах, из-за которых подобная терапия противопоказана пациентам с язвенной болезнью и язвой желудка, артериальной гипертензией, остеопорозом, катарактой и глаукомой. Отсутствие увеличения мышечной силы в течение 3 месяцев с момента начала лечения кортикостероидами говорит о стероидной резистентности воспалительной миопатии.
Альтернативными препаратами в лечении воспалительной миопатии являются цитостатики (азатиоприн, метотрексат, циклоспорин, циклофосфамид). Они применяются вместо кортикостероидов при наличии стероидной резистентности или в комбинации с ними для уменьшения дозы глюкокортикостероидов во избежание их побочных эффектов.
Поскольку системная воспалительная миопатия обычно имеет аутоиммунный характер, то в ее лечении зачастую применяется плазмаферез, позволяющий снизить количество ЦИК в крови.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Основные формы миопатии:
- I. Наследственные прогрессирующие мышечные дистрофии: Мышечная дистрофия Дюшенна и Беккера (Duchenne-strongecker), дистрофия Эмери-Дрейфуса (Emery-Dreifuss), фацио-скапуло-хумеральная, скапулоперонеальная, конечностно-поясная, дистальная форма, окулофарингеальная, прогрессирующая наружная офтальмоплегия. Конгенитальная мышечная дистрофия.
- II. Миопатии с миотоническим синдромом (мембранные миопатии).
- III. Воспалительные миопатии: полимиозит, СПИД, коллагенозы и др.
- IV. Метаболические миопатии (в том числе эндокринные и митохондриальные миопатии; миоглобулинемия и др.).
- V. Ятрогенные и токсические миопатии.
- VI. Алкогольная миопатия.
- VII. Паранеопластическая миопатия.

[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Где болит?
Мышечные дистрофии - термин, который применяют для обозначения наследственных форм миопатии, сопровождающихся дегенерацией мышц. Это - целая группа заболеваний, большинство из которых начинается в детском или юношеском возрасте, имеет неуклонно прогрессирующее течение и рано или поздно приводит к тяжёлой инвалидизации. Предложено несколько подробных классификаций мышечных дистрофий, построенных на разных принципах (генетических, биохимических, клинических), но унифицированнной классификации нет.
Дистрофин-дефицитные дистрофии объединяют в основном две формы: мышечную дистрофию Дюшенна (Duchenn) и мышечную дистрофию Беккера (strongecker).
Мышечная дистрофия Дюшенна, или псевдогипертрофическая мышечная дистрофия Дюшенна - самая злокачественная и самая распространённая форма Х-сцепленных миодистрофии. ферментемия (КФК) выявляется уже в неонатальном периоде, но клинические симптомы появляются в возрасте 2-4 лет. Эти дети поздно начинают ходить, им трудно или невозможно бегать и прыгать, они часто падают (особенно при попытке бежать), с трудом поднимаются по ступенькам или по наклонному полу (слабость проксимальных мышц) и ходят на больших пальцах (toe-walking) из-за контрактуры сухожилий стопы. Возможно снижение интеллекта. Характерна псевдогипертрофия икроножных мышц. Постепенно процесс принимает восходящее направление. Формируется гиперлордоз и кифосколиоз. К 8-10 годам грубо нарушается походка. Больной встаёт с пола с помощью характерных «миопатических «приёмов. К 14 - 15 годам больные, как правило, полностью обездвижены и умирают к 15-17 годам от слабости дыхательных мышц грудной клетки. ЭКГ обнаруживает нарушения почти в 90 % случаев (кардиомиопатия). Уровень КФК резко повышен. На ЭМГ - мышечный уровень поражения. В биоптате мышц неспецифические, хотя и характерные гистопатологические нарушения.
Мышечная дистрофия Беккера - вторая по частоте, но доброкачественная форма псевдогипертрофической миодистрофии. Начало заболевания между 5 и 15 годами. Паттерн вовлечения мышц такой же как при форме Дюшенна. Характерна слабость мышц тазового пояса и проксимальных отделов ног. Изменяется походка, появляются затруднения при вставании с низкого стула, при подъёме по лестнице. Развивается выраженная псевдогипертрофия икроножных мышц; процесс медленно распространяется вверх на мышцы плечевого пояса и проксимальных отделов рук. Уровень КФК повышен
Течение заболевание более благоприятное и медленное с более поздним нарушением трудоспособности.
Окулофарингеальная мышечная дистрофия характеризуется поздним началом (на 4-6 декаде жизни) и проявляется поражением глазодвигательных мышц, а также мышц глотки с нарушением глотания. Существует и форма с изолированным поражением только глазодвигательных мышц, которая, постепенно прогрессируя, приводит в конце концов к полной наружной офтальмоплегии. Последняя обычно протекает без двоения (окулярная миопатия, или прогрессирующая наружная офтальмоплегия Грефе). Диагноз подтверждается ЭМГ-исследованием. Уровень КФК поднимается редко (если процесс распространяется на другие поперечнополосатые мышцы).
Скапулоперонеальная (лопаточно-перонеальная) амиогрофия Давиденкова характеризуется прогрессирующей атрофией и слабостью в перонеальных группах мышц, а затем и в мышцах плечевого пояса. Некоторые исследователи считают, что синдром лопаточно-перонеальной атрофии является вариантом развития миодистрофий Ландузи-Дежерина.
Дистальная мышечная дистрофия является исключением из всей группы миодистрофий, так как поражает дистальную мускулатуру сначала голеней и стоп, затем - рук. Сухожильные рефлексы выпадают в той же последовательности Редко процесс распространяется на проксимальную мускулатуру. Для диагноза необходима сохранность чувствительности и нормальная скорость проведения возбуждения по нервам. Уровень КФК нормальный или слабо повышенный. ЭМГ подтверждает мышечный уровень поражения.
Существуют варианты дистальной мышечной дистрофии с началом в младенчестве, в детском возрасте, с поздним началом (тип Веландер), с накоплением десминовых включений.
Мышечная дистрофия Эмери-Дрейфуса имеет Х-сцепленный тип наследования, начинается в 4-5-летнем возрасте с характерным плече-перонеальным распределением атрофии и слабости (дистальные отделы остаются сохранными даже в далеко зашедших случаях). Типично раннее формирование контрактур в локтевых суставах, шее и в области ахилловых сухожилий. Другая типичная особенность - отсутствие псевдогипертрофий. Характерны нарушения ритма сердца, дефекты проведения (иногда полная блокада с внезапной смертью больного). Уровень КФК в сыворотке остаётся долгое время нормальным. ЭМГ показывает как нейрогенный, так и мышечный уровень поражения.
Особая группа - конгенитальные миопатии объединяет несколько заболеваний, которые выявляются обычно с рождения или в раннем детском возрасте и отличаются доброкачественным течением: часто они остаются стабильными на протяжении всей жизни; иногда они даже начинают регрессировать; если же отмечается в ряде случаев прогрессирование, то оно очень незначительное.
ЭМГ выявляет при этих формах неспецифические миопатические изменения. Мышечные фермены в крови либо нормальные либо слегка повышены. Диагноз ставится на основании электроно-микроскопического исследования.
Мембранные миопатии
Так называемые мембранные миопатии, куда относятся миотонические синдромы.
Воспалительные миопатии
В группу воспалительных миопатий включены такие заболевания как полиомиозит и дерматомиозит; миозит и миопатия с тельцами включения; миозит при заболеваниях соединительной ткани; саркоидная миопатия; миозит при инфекционных заболеваниях.
Наличие кожных изменений (эритемы, нарушения пигментации, телеангиоэктазии) является главным отличием дерматомиозита от полимиозита. Полиомиозит может быть первичным и вторичным (при злокачественном новообразовании).
Поражает чаще больных среднего или пожилого возраста (преобладают мужчины) и проявляется медленно прогрессирующей симметричной слабостью в конечностях. В отличие от других воспалительных миопатии здесь характерна как проксимальная, так и дистальная выраженная слабость мышц с вовлечением экстензоров стопы и флексоров пальцев рук. Боль не характерна. Иногда миозит с тельцами включения сочетается с заболеваниями соединительной ткани или иммунными нарушениями (болезнь Шегрена, тромбоцитопения). Уровень КФК умеренно повышен. ЭМГ обнаруживает смешанные нейрогенные и миопатические изменения в характере биоэлектрической активности. Биопсия мышц выявляет маленькие вакуоли с гранулами включения.
Такое сочетание особенно характерно для случаев смешанного (mixed) соединительнотканного заболевания. Оно характеризуется высокими титрами антирибонуклеопротеиновых антител; волчаночно-подобными высыпаниями на коже; изменениями соединительной ткани, напоминающими склеродермию; артритом и воспалительной миопатией. Клинически миопатия проявляется слабостью сгибателей шеи и мышц проксимальных отделов конечностей. Гистологически эта воспалительная миопатия напоминает дерматомиозит.
Воспалительная миопатия может наблюдаться при склеродермии, ревматоидном полиартрите, системной красной волчанке, синдроме Шегрена.
Может наблюдаться при саркаидозе (мультисистемное грануломатозное расстройство неизвестной этиологии). Грануломатозные изменения находят в оболочках мозга, головном мозге, гипофизе, спинном мозге и периферических нервах (а также в тканях глаза, в коже, лёгких, костях, лимфатических узлах и слюнных железах) Диагноз ставится на основания выявления мультисистемного поражения и биопсии мышц.
Бактериальные и грибковые миозиты встречаются редко и обычно являются компонентом системного заболевания. Паразитарные миозиты (токсоплазмоз, трихинеллёз, цистицеркоз) также встречаются нечасто. При цистицеркозе описана псевдогипертрофическая миопатия. Вирусные миозиты могут проявляться разной степенью тяжести от миалгии до рабдомиолиза. Разновидность таких воспалительных миопатии характерна для осложнений ВИЧ-инфекции и обычно наблюдается в контексте других неврологических и соматических проявлений СПИДа.
Метаболические миопатии
Метаболические миопатии включают карбогидратные миопатии, липидные миопатии, митохондриальные миопатии, эндокринные миопатии, миалгические синдромы, миоглобулинурию и токсические миопатии.
Карбогидратные миопатии обозначают как болезни накопления гликогена. Они связаны с недостаточностью тех или иных ферментов. Недостаточность мышечной фосфорилазы (болезнь Мак-Ардла) и других ферментов, а также липидные миопатии. Среди этих заболеваний осталась неупомянутой лизосомная болезнь накопления гликогена (болезнь Помпе - Ротре), проявляющаяся в первые месяцы жизни (быстро прогрессирующая мышечная слабость и массивная кардиомегалия) и приводящая к смерти на первом году жизни.
Синдром Кирнса-Сейра (Kearns-Sayre) проявляется прогрессирующей наружной офтальмоплегией. Он относится к спорадическим заболеваниям (но существует и семейный вариант прогрессирующей наружной офтальмоплегии) и, что типично, сопровождается вовлечением многих органов и систем. Болезнь начинается до 20-летнего возраста и проявляется пигментной дегенерацией сетчатки. Облигатные признаки этого заболевания: наружная офтальмоплегия, нарушения проводимости сердца и упомянутая пигментная дегенерация сетчатки. В качестве других дополнительных симптомов отмечают атаксию, ухудшение слуха, множественную эндокринопатию, увеличение содержание белка в ликворе и другие проявления. При семейном варианте прогрессирующей наружной офтальмоплегии возможны проявления слабости в мышцах шеи и конечностей.
Эндокринные миопатии встречаются при самых разнообразных эндокринных расстройствах. Довольно часто миопатия наблюдается при гипертиреозе. Слабость выявляется преимущественно в проксимальных отделах конечностей (редко - в дистальной и бульбарной мускулатуре) и подвергается обратному развитию при лечении гипертиреоза. Уровень КФК обычно не повышен. На ЭМГ и в мышечном биоптате - неспецифические миопатические изменения.
Однако встречаются случаи выраженного тиреотоксикоза, особенно при его быстром прогрессировании, сопровождающиеся рабдомиолизом, миоглобинурией и почечной недостаточноcтью. Слабость респираторых мышц, требующая механической вентиляции встречается редко.
Гипотиреоз часто сопровождается проксимальной мышечной слабостью, крампи, болью и ощущением напряжения (stiffness) мышц (хотя при объективном измерении слабость подтверждается нечасто). Эти симптомы исчезают при успешном лечении гипотиреоза. Мышечные гипертрофии редко встречаются при гипотиреозе, но их наличие у взрослых носит название синдрома Гофмана.
Кохер-Дебре-Семелайгна синдром наблюдается у детей (гипотиреоз с генерализованным мышечным напряжением и гипертрофией икроножных мышц). Уровень КФК повышен у 90 % больных гипотиреозом, хотя явный рабдомиолиз наблюдается очень редко. Миопатические изменения на ЭМГ вариируют от 8 % до 70 %. В мышечном биоптате - слабо выраженные признаки миопатии. Гипотиреоз ухудшает гликогенолиз в мышцах и окислительную способность митохондрий.
Мы не обсуждаем здесь дистиреоидную орбитопатию, также связанную с поражением мышечного аппарата орбиты.
Мышечная слабость, утомляемость и крампи очень часто сопутствуют болезни Аддисона. Иногда слабость может быть эпизодической. Здесь может иметь место периодический паралич с квадриплегией и гиперкалиемией.
Больные с гиперальдостеронизмом иногда отмечают приступы периодического паралича с гипокалиемией. 70 % этих больных жалуются на слабость.
На мышечную слабость часто жалуются больные с синдромом Иценко-Кушинга и больные, получающие длительное лечение глюкокортикоидами. Стреоидная миопатия чаще развивается медленно при длительном лечении таких, например, заболеваний, как системная красная волчанка, ревматоидный артрит, бронхиальная астма, полимиозит, и поражает преимущественно проксимально расположенные мышцы. Уровень КФК обычно не меняется; на ЭМГ - минимальные признаки миопатии.
Острая стероидная миопатия развивается реже: часто спустя неделю от начала лечения высокими дозами кортикостероидами. Такая миопатия может вовлекать респираторные мышцы. Острая стероидная миопатия может возникать и при лечении кортикростероидами больных миастенией.
Токсические миопатии
Токсические миопатии могу носить ятрогенный характер. Лекарственные препараты способны вызывать: миалгии, напряжение мышц (stiffness), или крампи; миотонию (задержку релаксации скелетных мышц после произвольного сокращения) - безболезненную проксимальную миопатию с мышечной слабостью; миозит или воспалительную миопатию; фокальную миопатию в области повреждения (инъекции); гипокалиемическую мимопатию при введении лекарств, вызывыающих гипокалиемию; митохондриальную миопатию в связи с торможением митохондриальной ДНК; рабдомиолиз (острый мышечный некроз с миоглобинурией и системными осложнениями).
Некротизирующая миопатия описана при использовании ловастатина (ингибитора синтеза холестерола), циклоспорина, аминокапроновой кислоты, прокаинамида, фенциклидина. Развивается мышечная слабость, боли (спонтанные и при пальпации мышц); уровень КФК повышается; на ЭМГ - картина миопатических изменений. Внутримышечное введение антибиотиков доксина ботулизма, хлорпромазина, фенитиона, лидокаина и диазепама может быть причиной локального мышечного некроза и фиброзной миопатии. Эметин вызывает прогрессирующую проксимальную миопатию. Такой же способностью обнаружена у клозапина, D-пеницилламина, гормона роста, интерферон-альфа-2b, винкристина.
Миалгию и мышечные крампи могут вызвать: ингибиторы ангиотензин-конвертирующего фактора, антихолинестераза, бета-адренергические агонисты, антагонисты кальция, отменам кортикостероидов, цитотоксические препараты, дексаметазон, диуретики, D-пеницилламин, левамизол, литий, L-триптофан, нифедипин, пиндолол, прокаинамид, рифампицин, сальбутамол. Лекарственно-индуцированная миалгия без мышечной слабости обычно быстро проходит после отмены препарата.
Алкогольная миопатия
Встречается в нескольких вариантах. Один тип характеризуется безболезненной, преимущественно проксимальной мышечной слабостью, развивающейся в течение нескольких дней или недель длительного злоупотребления алкоголем, что сочетается с выраженной гипокалиемией. Уровень печёночных и мышечных ферментов заметно повышается.
Другой тип алкогольной миопатии развивается остро на фоне длительного употребления алкоголя и проявляется выраженной болью и отёчностью мышц конечностей и туловища, что сопровождается симптомами почечной недостаточности и гиперкалиемией. Мионекроз (рабдомиолиз) отражается в высоком уровне КФК и альдолазы, а также миоглобинурией. Этому могут сопутствовать другие синдромы алкоголизма. Восстановление довольно медленное (недели и месяцы); характерны рецидивы, связанные с алкоголизмом.
Встречается вариант острой алкогольной миопатии, сопровождающийся тяжёлыми крампи и генерализованной слабостью. Возможна хроническая алкогольная миопатия, проявляющаяся безболезненной атрофией и слабостью мышц проксимальных отделов конечностей, особенно ног, с минимально выраженными знаками нейропатии.
[7], [8], [9], [10], [11]
Паранеопластическая миопатия
Отдельную позицию должна занимать миопатия с остеодистрофией и остеомаляцией, которая описана среди прочих паранеопластических синдромов.
Здесь не представлены некоторые редкие формы мышечных дистрофий, такик как миодистрофия Мэбри, миодистрофия Роттауфа-Мортье-Бейера, тазово-бедренная миодистрофия Лейдена-Мёбиуса, мышечная дистрофия Бетлема, дистальная миодистрофия Миоши.
Диагностика миопатий
Диагностические исследования при подозрении на миопатию включают, помимо клинического анализа, электромиоргафическое и электронейромиографическое исследование, анализ крови на ферменты (КФК, альдолаза, ACT, АЛТ, ЛДГ и др.). КФК в крови - наиболее чувствительный и достоверный показатель миодистрофического процесса. Исследуют также мочу на креатин и креатинин. Мышечная биопсия иногда незаменима для выявления природы миопатии (например, при конгенитальных миопатиях). Точная диагностика типа миопатии может потребовать проведения молекулярно-генетических, иммунобиохимических или иммуногистохимических исследований.
[12], [13], [14], [15], [16]
Существуют врожденные патологии, возникающие вследствие мутаций в генах. К одной из таких относится миопатия мышц. Заболевание отличается не до конца выясненными причинами развития, а также характерными проявлениями. Лечение патологии зависит от ее формы и выраженности признаков.
Что такое миопатический синдром
Миопатический синдром —, это поражение мышц человека. Для данной патологии свойственно хроническое течение с постоянным прогрессированием симптомов. При миопатическом синдроме происходит сбой в обмене веществ и самой структуре ткани мышц.
Итогом становится серьезная ограниченность в движениях человека, а также уменьшение силы в его мышцах.
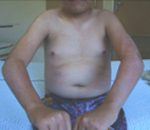
Под миопатическим синдромом подразумеваются различные патологии, связанные с поражением мышц. Они характеризуются дистрофическими явлениями в тканях. При этом чаще поражается скелетная мускулатура.
При синдроме возникает выборочная атрофия, затрагивающая некоторые волокна. Сама анимальная нервная система продолжает сохранять свою функциональность.
Причины
Основным провоцирующим фактором развития патологии выступает наследственная предрасположенность. Часто миопатия возникает у детей и подростков. Чем раньше проявился синдром, тем тяжелее он будет протекать у ребенка по мере взросления.
Возможные причины миопатии мышц ног и других частей тела также могут быть связаны с:
- осложнениями при хроническом тонзиллите,
- постоянными ОРВИ,
- перенесенным пиелонефритом,
- пневмонией бактериального типа,
- эндокринными заболеваниями (гипертиреоз и гипотиреоз),
- хроническим алкоголизмом,
- наркоманией,
- почечной и печеночной недостаточностью в хроническом проявлении,
- различными опухолями,
- сердечной недостаточностью,
- черепно-мозговыми травмами,
- повреждениями тазовых костей,
- сальмонеллезом.
Узнайте, как лечить слабость в мышцах рук и ног.
Болезнь может развиться из-за особенностей профессиональной деятельности на тяжелых и токсичных производствах. Возможным провоцирующим фактором для развития патологии также выступает постоянное перенапряжение.
Разновидности миопатий
Принято выделять следующие виды данной патологии:
- наследственные,
- метаболические,
- паранеопластические,
- воспалительные,
- токсические,
- мембранные.
Чаще встречаются наследственные миопатии. Они подразделяются на типы:
- Ювенильная, или миопатия Эрба. Ей подвержены молодые люди не старше 30 лет. Болезнь развивается в юношеском возрасте. Выражается атрофией бедренных мышц. Также при заболевании страдают мышцы в области тазового пояса. Характерными признаками патологии выступают «,утиная», походка и прекращение функционирования ротовых мышц.
- Миопатия Дюшенна. При ней значительно увеличивается мышечная масса, но сами мышцы остаются очень слабыми. По этой причине болезнь именуется псевдогипертрофической. Ей более подвержены мальчики. Патология отличается агрессивным течением, приводящим к инвалидности и гибели человека из-за осложнений на сердце и органах дыхания.
- Плече-лопаточно-лицевая миопатия. Заболеванию подвержены дети, начиная с 10 лет. В группе риска также юноши и девушки до 20 лет. Проявляется слабостью лицевых мышц. В дальнейшем атрофия наблюдается в области плечевого пояса и лопаток. Развивается гипертрофия глазных и ротовых мышц. Болезнь прогрессирует медленно и не всегда становится причиной инвалидности.
Миопатии воспалительного типа развиваются на фоне различных заболеваний. Это могут быть бактериальные (стрептококки), вирусные (грипп, ВИЧ, краснуха), паразитарные (токсоплазмоз) инфекции с поражением тканей мышц.

Метаболические миопатии развиваются из-за сбоя в липидном обмене. Данная разновидность мышечной атрофии также возникает на фоне нарушения обмена пуринов и гликогена.
Справка. Встречается также миопатия глаз. Болезнь протекает с единственным признаком —, близорукостью. Прочих нарушений у человека при патологии может не наблюдаться. В легкой форме у больного обычно протекает миопатия Беккера. При ней у молодых людей отмечается значительное увеличение объема икроножных мышц.
Характерные проявления болезни
Основным симптомом является слабость мышц при дистрофической миопатии. Она дополняется прогрессирующей утратой ими своих функций. К общим признакам патологии причисляются:
- частая утомляемость,
- боли в конкретных пораженных мышцах,
- нарушение суставной подвижности (ее снижение или повышение),
- ломота в мышцах,
- слабость в них и отсутствие силы.
Отдельные разновидности миопатии проявляются следующими признаками.
| Тип миопатии | Миопатия Эрба | Миопатия Дюшенна | Миопатия Беккера |
| Характерные симптомы | Дистрофия бедер, искривление в области позвоночника, атрофия спинных мышц, «,утиная», походка, утрата мышцами, расположенными возле рта, их прежней функциональности | Увеличение мышц на икрах, полная дистрофия большинства мышц тела, суставная деформация, проявления сердечной и дыхательной недостаточности | Усталость в ногах, мышечная атрофия в области таза |
При плече-лопаточно-лицевой миопатии симптомы выражаются в виде увеличения размера губ, нарушения произношения, невозможности закрыть глаза. У больного также наблюдается изменение мимики.
Диагностические мероприятия
Выявить заболевание можно только в ходе комплексного обследования пациента. Диагностика миопатии включает в себя:
- электронейрографию,
- электромиографию,
- изучение биохимического состава крови (исследование уровня креатинина),
- биопсию с забором мышечного лоскута.
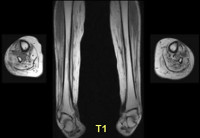
Дополнительно пациент может проходить рентген-исследование легких. Также при наличии проявлений сердечной недостаточности ему требуется проведение ЭКГ и УЗИ сердца. Обязательна консультация у кардиолога. Эффективным диагностическим методом для выявления миопатии также является МРТ.
Что делать, если потянул мышцу на шее?
Узнайте, что такое ригидность мышц.
Терапия
Полноценного лечения миопатии на данный момент еще не разработано. Терапия при различных формах патологии разрабатывается в рамках генной инженерии. Специалист изучает симптомы, и лечение миопатического синдрома у детей назначается с учетом их проявления.
В рамках симптоматической терапии показан прием витаминов. Основу лечения составляют витамины группы В и Е. Для улучшения обмена веществ в тканях мышц также назначаются аминокислоты. К ним относится глютаминовая кислота. Также рекомендован гидролизат, получаемый из свиного мозга.
Для нормализации метаболизма показаны аденозинтрифосфат, анаболические стероиды (деканоат нандролона), тиаминпирофосфат. Также при лечении используются неостигмин, галантамин, калиевые и кальциевые препараты. Назначаемые средства принимаются в комбинации. Курс терапии длится от 30 до 45 дней трижды в год.
Обязательно дополнение медикаментозного лечения физиотерапевтическими процедурами. Больной проходит следующие из них:
- лечение ультразвуком,
- электрофорез с использованием неостигмина,
- ионофорез с применением кальция.
Больным также показан легкий массаж. Им подбираются индивидуальные упражнения в рамках лечебной физкультуры. При этом упражнения рекомендуется делать в воде. Также пациентам требуется носить корректирующие корсеты. После консультации с ортопедом подбирается специальная обувь.
Важно! Для лечения миопатии Дюшенна разработан препарат «,Трансларна», (другое название —, «,Аталурен»,). Его регистрация разрешена в Европе в 2014 году.
Прогноз при болезни и ее профилактика
Поскольку для лечения патологии до сих пор не разработано радикальных средств, то прогноз для нее в целом неблагоприятен. Многое зависит от типа миопатии. Легкие формы заболевания прогрессируют медленно, поддерживающая терапия способствует улучшению качества жизни больного.
Тяжелее протекают миопатии, развившиеся в раннем детстве. Они в большинстве случаев приводят к инвалидности. Неблагоприятный прогноз и для миопатии Дюшенна. При ней поражаются сердечная мышца и органы дыхания. Заболевание прогрессирует быстрее, чем другие его разновидности, и нередко приводит к летальному исходу от сердечной или дыхательной недостаточности.
Прогноз при миопатии вторичного типа оценивается как благоприятный. При этом важно, чтобы было успешно вылечено основное заболевание.

Главным способом профилактики является полное лечение любых инфекционных болезней. Также обязательна своевременная терапия эндокринных нарушений. Необходимо избегать воздействия на организм токсических веществ и стараться корректировать сбои в обмене веществ.
Внимание! Семейным парам, которые планируют зачатие ребенка, рекомендуется проходить консультацию у генетика на предмет возможной предрасположенности к развитию заболевания.
Заключение
Миопатия представляет собой группу хронических патологий разной степени прогрессирования, при которых развивается мышечная атрофия. Болезнь может быть как первичной, так и вторичной. Основной ее причиной появления считается генетическая предрасположенность. Тяжесть протекания патологии зависит от ее вида.
Наиболее тяжелые последствия для организма имеет миопатия сердечной мышцы. Заболевание провоцирует развитие ее недостаточности, что чревато смертельным исходом для больного. Из-за хронического статуса миопатии и отсутствия радикальных препаратов для ее лечения данная патология по факту является неизлечимой.
Читайте также: