
Доброкачественная пароксизмальная кривошея детей раннего возраста
Доброкачественный младенческий пароксизмальный тортиколлис (ДМПТ) - относительно редкое функциональное расстройство у детей первого года жизни, представленное повторяющимися с определенной регулярностью стереотипными дистониями мышц шеи (тортиколлис), что визуально проявляется непроизвольным наклоном, поворотом и приведением головы к плечу и удержанием некоторое время соответствующей позы.
Доброкачественный младенческий пароксизмальный тортиколлис (ДМПТ) как самостоятельная нозологическая единица впервые был описан C.Snyder на основании наблюдений за 12 детьми раннего возраста (до сих пор наиболее обширная и репрезентативная когорта наблюдаемых детей).
Эпидемиология доброкачественного младенческого пароксизмального тортиколлиса (ДМПТ)
Истинная частота встречаемости доброкачественного младенческого пароксизмального тортиколлиса (ДМПТ) не установлена. В настоящее время имеется порядка 120 документированных описаний данного моторного феномена у младенцев (Campos-Castello J., Rosman N. et al., Cuvellier J.-C. и et al.). Однако не исключено, что многие случаи остаются нераспознанными либо ошибочно трактуются в структуре иных патологических состояний (Rosman N. et al.).
Следует признать, что, анализируя истории болезни детей первых двух лет жизни, пролеченных в неврологических отделениях ДГБ №4 святой Ольги Санкт-Петербурга за последние 3 года, возможно, мы и наблюдали, но не диагностировали ни одного случая ДМПТ.
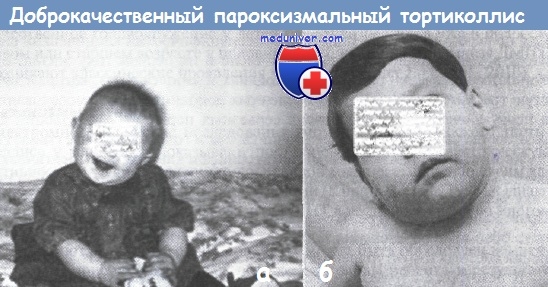
Характерная поза ребенка при доброкачественном пароксизмальном тортиколлисе
(наклон и приведение головы, незначительное приподнимание плеча на одноименной стороне и легкая ротация лица в противоположную сторону)
В исследовании было показано, что у 198 (94,2%) из них истинная причина спонтанно остро развившегося тортиколлиса так и осталась неизвестной. Автор высказал ряд гипотез происхождения данного состояния, в основном делая акцент на структурно-функциональных расстройствах шейного отдела позвоночника и атланто-окципитального сочленения, что, в общем, и понятно, учитывая хирургическую направленность отделения.
Были ли среди наблюдаемых автором дети с ДМПТ, особенно принимая во внимание значительно более старший возраст пациентов, отсутствие полноценной неврологической оценки состояния и отдаленного проспективного наблюдения, не совсем ясно. Это клиническое исследование, возможно, отчасти расширяет представления о возрастных рамках дебюта ДМПТ и, безусловно, требует дальнейшего более детального изучения и осмысления полученных результатов.

Механизмы развития доброкачественного младенческого пароксизмального тортиколлиса (ДМПТ)
Электромиографические исследования в момент идиопатического тортиколлиса у младенцев показывают постоянную мышечную электрическую активность в грудино-ключично-сосцевидной мышце, схожую по своим характеристикам с дистониями иной этиологии (Kimura S. et al.). Однако непосредственные механизмы развития ДМПТ в настоящее время во многом остаются неизвестными.
Полагают, что тортиколлис при доброкачественном младенческом пароксизмальном тортиколлисе (ДМПТ) может быть обусловлен компенсаторным, приспособительным положением головы ребенка в результате функционального нарушения периферического отдела вестибулярного аппарата (лабиринтит) (Snyder С., Eviatar L.). Другие исследователи рассматривают транзиторную дисфункцию центральных структур вестибулярной системы или вестибуломозжечковых связей как основную причину ДМПТ (Sanner G. et al., Deonna Т. et al.).
Существуют предположения, что при ДМПТ у детей отмечается функциональная незрелость мозжечково-стволовых связей и/или нейротрансмиттерных систем (Aker P. et al.). B.John и соавт. в эксперименте показали снижение метаболизма глюкозы в мозжечке и подкорковых ганглиях в совокупности с транзиторной сосудистой гипоперфузией коры височных долей и базальных отделов мозга, что может приводить к состояниям, напоминающим ДМПТ.
Аннотация научной статьи по клинической медицине, автор научной работы — Окунева И.В., Бобытова М.Ю., Какаулина В.С., Нестеровский Ю.Е.
Доброкачественный пароксизмальный тортиколис младенчества редкое состояние, характеризующееся приступами кривошеи, которые продолжаются от нескольких часов до нескольких недель, возникают у здоровых детей и самостоятельно купируются. Предположительно данное состояние является предшественником мигрени и связано с мутациями в генах CACNA 1A и PRRT2. Дифференциальный диагноз доброкачественного пароксизмального тортиколиса необходимо проводить, прежде всего, с эпилепсией. Статья включает краткий обзор литературы и описание 6 случаев (собственное наблюдение авторов).
Похожие темы научных работ по клинической медицине , автор научной работы — Окунева И.В., Бобытова М.Ю., Какаулина В.С., Нестеровский Ю.Е.
BENIGN PAROXYSMAL TORTICOLLIS OF INFANCY
Benign paroxysmal torticollis of infancy a rare condition, characterize by episodes of torticollis with duration from several hours to several weeks, which appears in healthy children and is self-limited. It is proposed that this condition is predictor of migraine and connected with mutations in CACNA 1A and PRRT2. Differential diagnosis of benign paroxysmal torticollis includes, first of all, epilepsy. The article includes a short review and own description of 6 cases.
НАБЛЮДЕНИЯ ИЗ ПРАКТИКИ
ДОБРОКАЧЕСТВЕННЫЙ ПАРОКСИЗМАЛЬНЫЙ ТОРТИКОЛИС МЛАДЕНЧЕСТВА
И.В. Окунева1, М.Ю. Бобылова2, B.C. Какаулина23, Ю.Е. Нестеровский4
BENIGN PAROXYSMAL TORTICOLLIS OF INFANCY
I.V. Okuneva1, M.Yu. Bobylova2, V.S. КакаиНпа2-3, Yu.E. Nesterovsky4
1 —Морозовская детская городская клиническая больница, Москва;
2 — Институт детской неврологии и эпилепсии им. Святителя Луки, Москва;
3 — Российская детская клиническая больница, Москва;
4 — Кафедра неврологии, нейрохирургии и медицинской генетики педиатрического факультета РНИМУ, Москва.
Доброкачественный пароксизмальный тортиколисмладенчества - редкое состояние, характеризующееся приступами кривошеи, которые продолжаются от нескольких часов до нескольких недель, возникают у здоровых детей и самостоятельно купируются. Предположительно данное состояние является предшественником мигрени и связано с мутациями в генах CACNA 1A и PRRT2. Дифференциальный диагноз доброкачественного пароксизмального тортиколиса необходимо проводить, прежде всего, с эпилепсией. Статья включает краткий обзор литературы и описание 6 случаев (собственное наблюдение авторов).
Ключевые слова: доброкачественный пароксизмальный тортиколис, мигрень, кривошея, пароксиз-мальные неэпилептические состояния.
Benign paroxysmal torticollis of infancy - a rare condition, characterize by episodes of torticollis with duration from several hours to several weeks, which appears in healthy children and is self-limited. It is proposed that this condition is predictor of migraine and connected with mutations in CACNA 1A and PRRT2. Differential diagnosis of benign paroxysmal torticollis includes, first of all, epilepsy. The article includes a short review and own description of 6 cases.
Key words: Benign paroxysmal torticollis, migraine, torticollis, paroxysmal non-epileptic condition.
Дмальный тортиколис (ДПТ) - редкое состояние, характеризующееся повторными приступами кривошеи у детей раннего возраста. Кривошея может сопровождаться наклоном головы в сторону (при последующих приступах возможно чередование сторон) с поворотом головы или без него. В момент приступа голову ребенка можно пассивно вернуть в среднее положение. Эпизод кривошеи сопровождается бледностью, беспокойством, дурнотой, рвотой и атаксией (если ребенок уже научился ходить). Иногда во время приступа ДТП отмечаются другие дистони-ческие феномены, например, ретроко-лис или асимметричное изменение позы таза [5], а также такие симптомы как заведение глаз вверх, сгибание верхних конечностей, птоз и мидриаз на одноименной стороне [3]. ДПТ возникает у младенцев на первом году жизни, длится от нескольких часов до нескольких суток и сменяется спонтанными ремис-
сиями. В периодах между приступами ребенок неврологически здоров. ДПТ проходит бесследно или трансформируется в доброкачественное пароксиз-мальное головокружение детского возраста, синдром циклических рвот или мигрень. ДПТ впервые было описано С. Snyder в 1969 г.
ТОМ VIII ВЫПУСК 2 2013
ких синдромов детства. Тем не менее, ДПТ считают предшественником мигрени [10].
ДПТ встречается крайне редко, отчего, как правило, вызывает диагностические трудности. Распространенность неизвестна из-за отсутствия достоверных статистических данных. По данным Al-Twaijri W.A., Shevell M.I. (2002), ДПТ встречается у 1 на 500 детей грудного возраста, причем у 10% этих детей в дальнейшем формируется мигрень [1].
Этиология и патогенез в настоящее время не известны. Существует несколько гипотез.
1. Функциональная незрелость вестибулярной системы. В наблюдении T. Deonna, D. Martin (1981) в 9 из 12 случаев отмечалась положительная калориметрическая проба (возникал нистагм при раздражении слухового прохода холодной водой). В более старшем возрасте ДПТ трансформируется в такие состояния, как доброкачественное паро-ксизмальное головокружение, абдоминальная мигрень, синдром циклической рвоты, что позволяет расценивать ДПТ как предшественник мигрени [8].
2. Каналопатия. В настоящее время установлено, что семейные случаи ДПТ связано с двумя мутациями - CACNA1A и PRRT2. Мутация гена CACNA1A вызывает нарушение функции кальциевого канала и приводит к нескольким паро-ксизмальным состояниям [14]. У части больных с семейной гемиплегической мигренью и атаксией с диагностированной мутацией в гене CACNA1A в раннем возрасте отмечались такие феномены, как доброкачественная девиация глаз вверх и доброкачественный торти-колис [17, 11]. Недавно открыта мутация гена PRRT2, встречающаяся при таких доброкачественных состояниях, как пароксизмальная кинезиогенная дискинезия, доброкачественная семейная инфантильная эпилепсия, доброкачественный пароксизмальный торти-
колис, гемнплегнческая мигрень с началом в детском возрасте. Доказано, что данные виды пароксизмальных расстройств реагируют на препараты карба-мазепина, дозы и продолжительность приема которого зависит от эпилептической или неэпилептической природы пароксизмов [7]. Ген РИИТ2 кодирует трансмембранный протеин 2, содержащейся в основном в клетках церебральной коры и базальных ганглиев. Функция данного белка до конца не изучена, однако, считается, что он играет существенную роль в мембранном транспорте ионов, участвуя в работе синапсов корково-подкорковых связей [4, 12]. Предположительно нарушение функции трансмембранного протеина 2 приводит к повышению возбудимости коры и базальных ядер. Однако пока нет объяснения, почему в результате этой мутации возникают столь разные клинические состояния, характеризующиеся разным возрастом дебюта [16].
Клинические проявления. Общая клиническая картина ДПТ хорошо известна, но большинство публикаций представляет единичные наблюдения. Наибольшее число случаев (75 случаев) описано Б.^ БазЬеег [2]. По данным этого автора, в большинстве случаев ДПТ возникает спорадически, лишь у 6% пациентов прослеживается семейный анамнез. Тем не менее, у 50% детей с ДПТ семейный анамнез отягощен по мигрени [2]. По данным Р. Drigo и соавт. (2000) [9], которые наблюдали 18 детей с пароксизмальным тортиколисом в течение 10 лет, оказалось, что у 6 детей (33%) состояние трансформировалось в мигрень. В выборке Р. Drigo тортико-лис сочетался с тортипелвисом в 54%, приступ начинался утром после пробуждения (50%), почти у половины детей пароксизмы длились больше 7 дней, в 100% ДПТ самостоятельно купировался к 3 годам [9]. В 95% случаев заболевание дебютирует в возрасте от 2 до 8 мес., причем у 50% пациентов - в первые 3 месяца жизни. Приступы возникают преимущественно в утренние часы без явных провоцирующих факторов или на фоне прорезывания зубов, сопутствующих инфекционных заболеваний, при смене положения тела. Возникновению непосредственно кривошеи может предшествовать продромальное состояние в виде беспокойст-
НАБЛЮДЕНИЯ ИЗ ПРАКТИКИ
ва, раздражительности, ярких вегетативных реакций. Приступы кривошеи длятся до 1 суток - у 30% детей, несколько суток - у 30%, и несколько недель - у 30%. Но выраженность кривошеи по мере продолжения приступа постепенно уменьшается. Повторные эпизоды возникают 1 раз в несколько недель - несколько месяцев, причем с возрастом продолжительность приступов укорачивается, а промежуток между ними увеличивается. К 3 годам ДПТ всегда купируется. У части детей в дошкольном возрасте дебютируют такие пароксиз-мальные состояния, как доброкачественное головокружение либо мигрень.
ДПТ является диагнозом исключения. Необходим тщательно собранный анамнез и клинический осмотр ребенка. Дополнительные методы диагностики включают нейросонограмму с допп-лерографией брахиоцефальных сосудов, нейроофтальмоскопическое исследование и ЭЭГ в момент приступа кривошеи. По показаниям, при соответствующем дифференциальном диагнозе, требуется проведение МРТ, аудиомет-рии, рентгенографии шейного отдела позвоночника, обследования у гастроэнтеролога. Окончательный диагноз чаще всего возможен только после повторного приступа пароксизмальной кривошеи [6].
Дифференциальный диагноз следует проводить с гастроэзофагеальным реф-люксом (синдром Сандифера), идиопа-тической торсионной дистонией и эпилепсией; необходимо также исключать наследственную и приобретенную патологию задней черепной ямки и краниоцервикального перехода. Будучи пароксизмальным состоянием по своей природе, ДПТ часто требует исключения наследственных болезней обмена (в частности, митохондриаль-ных заболеваний) и транзиторных ишемических атак [6].
Специфического лечения не существует. Основной врачебной помощью
ТОМ VIII ВЫПУСК 2 2013
рые чудесным образом излечивались отменой ранее назначенной терапии.
В случае тошноты и многократной рвоты возможно применение противо-рвотных препаратов. Некоторые авторы советуют привентивное применение ципрогептадина для предотвращения приступов ДПТ у детей [10, 15].
Прогноз ДПТ достаточно благоприятный; у большинства детей приступы ДПТ купируются к 3 годам жизни. Однако возможна трансформация ДПТ в мигрень в более старшем возрасте.
Представляем собственное описание - 6 случаев ДПТ (таблица 1). В нашей небольшой выборке можно отметить равное соотношение по полу, де-
Пациент, возраст Пол Дебют Провоцирующий фактор Клинические проявления острого периода Дифференциальный диагноз Семейный анамнез
Д, 2 мес. Жен 1 мес. Массаж Поворот головы вправо, отведение глаз вправо, гипомимия, нистагм при сохранном сознании и двигательной активности в конечностях. При попытке пассивного поворота головы влево - негативная реакция. В дальнейшем при купании отмечался поворот таза влево. Длительность эпизода - 2 ч. Последствие перинатального поражения ЦНС Мигрень у матери
Р, 2 года Жен 2 мес. Нет Приступы кривошеи слева, сопровождавшиеся вялостью, повторной рвотой, после 9 мес. - падением влево. Объемное образование ЗЧЯ Данных нет
Е, 3 года Муж 4 мес. Нет Насильственный поворот головы вправо, длительностью несколько часов, последний эпизод в 8 мес. Эпилепсия Данных нет
М., 1 год Муж 6 мес. Введение прикорма Кормление в положении сидя на стуле провоцировало кривошею. Первый эпизод длился 3 недели, затем отмечалось еще несколько эпизодов длительностью несколько дней. Эпилепсия Мигрень у отца
В., 9 мес. Жен 3 мес. Появление колик и лечение лак-тазной недостаточности Кривошея в течение нескольких часов Гастроэзо- фагеальный рефлюкс Не отягощен
М, 1 год 9 мес. Жен 3 мес. На фоне ринита, после сна Наклон головы вправо, заведение глаз влево, сначала плакала, затем засыпала, но могла просыпаться, отвечать на вопросы, следить за предметами и поворачивать голову вправо и влево, при этом наклон головы к плечу сохранялся, продолжительность - 2 ч. Болезнь Гризеля Мигрень у матери и бабушки
Таб. 1. Течение доброкачественного пароксизмального тортиколиса, 6 случаев.
бют 1-го приступа в первые 6 мес., отсутствие достоверных провоцирующих факторов. Все дети были госпитализированы и обследованы в стационаре, дифференциальный диагноз представлен в таблице. Обследование было проведено оперативно, однако, в большинстве случаев приступ закончился раньше, чем была закончена диагностика. Характерной особенностью является поведение родителей - паническое в момент дебюта приступа и негативное по отношению к врачам после выздоровления. Когда приступ кривошеи проходит, родители понимают, что ребенок здоров, они начинают категорически отказываться от дальнейших диагностических процедур. В связи с этим собрать катамнестические сведения сложно. Без сомнения дети с ДПТ после обследования в стационаре должны наблюдаться неврологом амбулаторно в декретируемые сроки, для выяснения прогноза. В таких случаях (как и во многих других) очень важна преемственность и обратная связь между врачами стационара и поликлиники, однако, данная тема слишком глобальна и выходит за рамки нашего небольшого обзора.
РУССКИЙ ЖУРНАЛ ДЕТСКОЙ НЕВРОЛОГИИ
НАБЛЮДЕНИЯ ИЗ ПРАКТИКИ
Обсуждение. В литературе ДПТ описан достаточно давно, однако, до сих пор вызывает диагностические трудности у практикующих врачей-неврологов. Нередко проводится большое количество исследований и процедур, в том числе инвазивных, назначается лечение, в котором нет необходимости. Мы считаем, что при возникновении первого эпизода ДПТ необходимо проведение нейровизуализации для исключения опухоли задней черепной ямки; также большое диагностическое значение имеет видео-ЭЭГ мониторинг, позволяющий исключить эпилептическую природу состояния. Детям с ДПТ должно быть проведено генетическое исследование - поиск мутаций в генах СЛСКЛ1Л и РИИТ2. По нашему мнению, специфического лечения ДПТ не требует, однако, в ряде случаев показана симптоматическая терапия во время приступов.
ДПТ ждет дальнейшего изучения, особенно в вопросах катамнестическо-го наблюдения для определения прогноза и взаимосвязи данного состояния с мигренью и ее предикторами.
1. Al-Twaijri W.A., Shevell M.I. Pediatric migraine equivalents: occurrence and clinical features in practice // Pediatr Neurol. - 2002. - V. 26. - P. 365-368.
2. Basheer S.N. Paroxysmal torticollis // Journal of Pediatric Neurology. - 2010. - V. 8. - P. 69-71.
3. Cataltepe S.U., Barron T.F. Benign paroxysmal torticollis presenting as "seizures" in infancy // Clin Pediatr (Phila). - 1993. - V. 32. - P. 564-5.
4. Chen W.J., Lin Y., Xiong Z.Q., et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia // Nat Genet. - 2011. - V. 43. - P. 1252-5.
5. Chutorian A.M. Benign paroxysmal torticollis, tortipelvis and retrocollis of infancy // Neurology. - 1974. -V. 24. - P. 366-7.
6. Cuvellier J-C, Lepine A. Childhood periodic syndromes // Pediatr Neurol. - 2010. - V. 42. - P. 1-11.
7. Dale R.C., Gardiner A., Antony J., Houlden H. Familial PRRT2 mutation with heterogeneous paroxysmal disorders including paroxysmal torticollis and hemiplegic migraine // Dev Med Child Neurol. - 2012. - V. 54(10). -P. 958-960.
8. Deonna T., Martin D. Benign paroxysmal torticollis in infancy // Arch Dis Child. - 1981. - V. 56(12). - P. 956-959.
ТОМ VIII ВЫПУСК 2 2013
9- Drigo P., Carli G., Laverda A.M. Benign paroxysmal torticollis of infancy // Brain & Development. - 2000. -V. 22. - P. 169-172.
10. Gelfand A.A. Migraine and childhood periodic syndromes in children and adolescents // Curr Opin Neurol. -2013. - V. 26(3). - P. 262-8.
11. Giffin N.J., Benton S., Goadsby P.J. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation // Dev Med Child Neurol. - 2002. - V. 44. - P. 490-493.
12. Heron S.E., Grinton B.E., Kivity S., et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome // Am J Hum Genet. - 2012. - V. 90. - P. 152-60.
13. Kimura S., Nezu A., Electromyographic study in an infant with benign paroxysmal torticollis // Pediatric neurology. - 1998. - V. 19. - P. 236-238.
14. Kors E.E., Melberg A., Vanmolkot K.R., et al. Childhood epilepsy, familial hemiplegic migraine, cerebellar ataxia, and a new CACNA1A mutation // Neurology. - 2004. - V. 28;63(6). - P. 1136-7.
15. Lewis D.W., Gozzo Y., Avner M., Yonker M., Landy S.H. Primary headache disorders in children, adolescents, and young adults / In: Winner P., Lewis D.W., editors. Young adult and pediatric headache management. -Hamilton. Ontario, Canada: B.C. Decker, 2005. - P. 41-115.
16. Meneret A., Gaudebout C., Riant F., et al. PRRT2 mutations and paroxysmal disorders // Eur J Neurol. - 2013 [Epub ahead of print]
17. Roubertie A., Echenne B., Leydet J. et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family // J of Neurology. - 2008. - V. 255. - P. 1600-1602.
Медицинский эксперт статьи

- Где болит?
- Что нужно обследовать?
- Как обследовать?
Пароксизмальные дискинезии - это полиэтиологическое заболевание, проявляющееся приступами дистонических (а также хореических, миоклонических и баллистических) движений и патологических поз без потери сознания. До сих пор не создано унифицированной классификации этих приступов. В качестве классификационных критериев используют: время суток, в которое наблюдаются приступы (дневные - ночные), провоцирующие факторы (кинезиогенные - некинезиогенные), длительность приступа (короткие - длительные), наследственность (семейные - приобретённые или первичные - вторичные).
Основные клинические формы пароксизмальных дискинезии:
- Пароксизмальная кинезиогенная дискинезия.
- Пароксизмальная некинезиогенная дискинезия.
- Пароксизмальная дискинезия, индуцированная физической нагрузкой.
- Пароксизмальная гипногенная дискинезия.
- Доброкачественный пароксизмальный тортиколлис у младенцев.
- Пароксизмальные дискинезии в картине альтернирующей гемиплегии у детей.
- Психогенные гиперкинезы пароксизмального характера.
Пароксизмальная кинезиогенная дискинезия
Первичная (наследственная и спорадическая) кинезиогенная дискинезия начинается в 80 % случаев в возрасте от 8 до 17 лет (возможны вариации от 1 года до 30 лет и старше), чаще встречается у мужчин и проявляется короткими приступами (в большинстве случаев меньше 1 минуты) насильственных движений. Характерна высокая частота приступов: почти все больные страдают от ежедневных единичных приступов; у многих они возникают несколько раз в день, а в период обострения - до 100 в день и чаще. Одна из отличительных черт пароксизмальной кинезиогенной дискинезии - провокация приступов движением. Обычно это внезапное неподготовленное автоматически совершаемое движение. Испуг и вздрагивание также могут провоцировать приступ. Пароксизм развивается на той стороне тела, на которой совершалось движение (обычно рукой или ногой). Приступ, начавшись в руке (или ноге) может распространяться по гемитипу или (реже) ограничивается одним регионом тела или даже его частью. У одного и того же пациента могут от приступа к приступу чередоваться левосторонние, правосторонние и двусторонние приступы. В моторном рисунке приступа доминируют тонические и дистонические, реже другие, движения и позы.
Непосредственно перед приступом большинство больных испытывают сенсорную ауру в виде ощущения стягивания, покалывания, онемения, скованности, мурашек в той конечности, которая будет вовлечена в пароксизм. В случае билатеральных приступов аура чаще двусторонняя. Некоторые больные сообщают о возможности определённого контроля над приступами: чувствуя приближение приступа некоторые пациенты могут предотвратить его, полностью прекратив все движения или удерживая поражённую конечность другой рукой. Иногда приступ удаётся предотвратить медленным выполнением движения, превращая его из автоматического в высоко контролируемое. Практически все больные сообщают о рефрактерном периоде, когда в течение короткого времени после приступа (как правило 5-20 минут) никакие провоцирующие стимулы не способны вызвать приступ. Типична сохранность сознания в приступе и отсутствие постприступной спутанности. Неврологический статус во время приступа и в межприступный период без отклонений от нормы.
Пароксизмальная некинезиогенная дискинезия
Первичная (наследственная и спорадическая) некинезиогенная дискинезия начинается почти исключительно в детстве (в двух третях случаев дебют заболевания приходится на возраст раньше 5 лет), среди заболевших преобладают лица мужского пола. Эта форма характеризуется более редкими приступами (1 раз в неделю или 2-3 раза в месяц). Сами приступы более длительны: от 5 минут до 4-5 часов и больше. Во взрослой жизни отмечается тенденция к спонтанному улучшению. Приступы развиваются либо спонтанно, либо провоцируются алкоголем, кофе, анальгетиками, стрессом, менструациями и другими факторами. Здесь также характерна сенсорная аура и частичный контроль над приступами (чаще всего с помощью релаксации). Двигательный рисунок приступа практически такой же как при кинезиогенной дискинезии.
Пароксизмальная дискинезия, индуцированная физической нагрузкой
Пароксизмальная гипногенная дискинезия
Все вышеуказанные варианты пароксизмальных дискинезии относятся к первичным (наследственным или спорадическим) формам. ЭЭГ и неврологический статус в межприступном периоде обычно не обнаруживают отклонений от нормы. ЭЭГ во время приступа зарегистрировать трудно из-за артефактов, связанных с движениями (дискинезиями). Вторичные (симптоматические) формы вышеупомянутых дискинезии описаны при многих заболеваниях. К ним относятся: детский церебральный паралич, рассеянный склероз, гипопаратиреоз, псевдогипопаратиреоз, гипогликемия, тиреотоксикоз, инфаркт мозга (в том числе при системной красной волчанке), транзиторные ишемические атаки, кровоизлияние в продолговатый мозг, артерио-венозная мальформация, черепно-мозговая травма, энцефалит (в том числе при ВИЧ-инфекции), ятрогенные (церукал, метилфенидат) и токсические (кокаин, алкоголь) формы. Здесь возможны более разнообразные изменения ЭЭГ и неврологического статуса. При всех вышеупомянутых формах пароксизмальных дискинезий отмечается лечебный эффект антиконвульсантов.
Доброкачественный пароксизмальный тортиколлис у младенцев
Доброкачественный пароксизмальный тортиколлис у младенцев встречается ещё реже и развивается, как видно из названия, только у младенцев грудного возраста. Заболевание возникает в первые месяцы жизни и проявляется повторяющимися эпизодами подёргиваний головой и кривошеи длительностью от 15 минут до нескольких часов. Эти эпизоды иногда сопровождаются тошнотой, рвотой и атаксией. Атаки повторяются ежемесячно и спонтанно прекращаются в ближайшие годы. Характерна генетическая предиспозиция к мигрени. У многих больных с доброкачественным пароксизмальным тортиколлисом в дальнейшем развивается мигрень. ЭЭГ и калорическая проба во время приступа тортиколлиса обычно показывают нормальную картину.
Пароксизмальные дискинезий в картинеальтернирующей гемиплегии у детей
Альтенирующая гемиплегия у детей относится к редким заболеваниям и характеризуется: дебютом болезни в возрасте до 3-х лет (иногда в возрасте 3 месяцев); повторными атаками гемиплегии (с чередованием поражённой стороны тела) длительностью от нескольких минут до нескольких дней; наличием других пароксизмальных феноменов (дистонии, хореи, нистагма, вегетативных нарушений в виде тахикардии, мидриаза и гипергидроза во время гемиплегии или независимо от неё); эпизодами билатеральной гемиплегии; улучшением во время сна и прогрессирующим ухудшением неврологических и психических функций.
Характерна задержка психического развития. Неврологический статус характеризуется ступенеобразным ухудшением, так как восстановление функций после отдельных приступов может быть неполным. Самыми частыми симптомами являются дистония, спастичность, псевдобульбарный паралич и атаксия. МРТ обнаруживает прогрессирующую атрофию червя мозжечка. Большинство случаев (кроме одной семьи) относятся к спорадическим.
Дифференциальный диагноз проводят с пароксизмальными дискинезиями, гемиплегической мигренью, эпилепсией, инсультом, допареспонсивной дистонией (дистонией, чувствительной к дофамину).
Психогенные гиперкинезы пароксизмального характера
В целом любой психогенный гиперкинез отличается от органического по четырём факторам: двигательным рисунком, динамикой гиперкинеза, синдромальным окружением и течением заболевания. Для обоснованного диагноза важна позитивная диагностика психогенного ("невротического") расстройства и исключение классических форм органических гиперкинезов. В настоящее время разработаны критерии диагностики психогенного тремора, психогенного миоклонуса, психогенного паркинсонизма, психогенной дистонии, а также критерии диагностики сочетания психогенных и органических гиперкинезов; сформулированы критерии доказанных (документированных), достоверных, вероятных и возможных психогенных двигательных расстройств. Однако их изложение выходит за рамки этого раздела книги.

[1], [2], [3]
Кривошея очень распространена среди маленьких детей, она регистрируется примерно у 16% новорожденных. Однако при раннем выявлении, аномалию можно исправить даже без хирургического вмешательства.
Итак, как вовремя заметить и решить проблему?
Определение патологии
Кривошея приводит к заметному повороту и наклону головы младенца, что зачастую провоцирует вынужденное положение головы и шеи, а также деформацию части черепа (особенно в области нижней челюсти).
Мышечная кривошея может быть врожденной, как порок развития мышечных волокон, или возникает из-за болезней.
В любом случае, развитие кривошеи приводит к сокращению длины мышцы шеи, вызывая ее деформацию, поворот головы, ее наклон в сторону. В итоге, головка ребенка фиксируется в таком неправильном, порочном положении, пока не будет проведено медицинское вмешательство.
Что такое кривошея?
Кривошея — аномальный поворот головы и шеи из-за укорочения грудино-ключично-сосцевидной мышцы (ГКС-мышца).
- От области грудины (грудной кости).
- Основания ключицы.
- Она крепится к височной кости черепа прямо под ушами.
ГКС-мышца имеется на обеих сторонах головы и шеи, управляя поворотами головы и сгибанием шеи в разных направлениях. Когда одна мышца сокращается или недоразвита, она тянет шею в своем направлении, в то время как ГКС-мышца с другой стороны чрезмерно растягивается и перенапрягается.
При кривошее, ГКС-мышца сокращается и остается в таком состоянии постоянно, в силу чего возникает скручивание шеи на одну сторону. Отсюда и название torticollis (кривошея по латыни), что означает “скрученная шея”.
В зависимости от стороны, на которую повернута голова ребенка, кривошея может называться “правосторонняя кривошея”, “левосторонняя” или “позиционная кривошея”.
Младенцы чувствуют только дискомфорт, вызванный аномальным положением шеи при лежании.
Что вызывает кривошею у младенцев – причины патологии
- Врожденная мышечная кривошея.
- Приобретенная кривошея.
Обсудим с вами подробнее причины развития каждого типа мышечной кривошеи у малышей.
Врожденная кривошея, называемая также врожденной грудино-ключично-сосцевидной мышечной аномалией, выявляется, когда ребенок рождается с явным и выраженным искривлением шеи. Либо же она развивается в первые три месяца его жизни.
- Рождение в ягодичном предлежании. Если малыш рождается в ягодичном предлежании, при аномалиях родового акта шея ребенка может застрять в родовом канале. В результате, при оказании акушерского пособия она может растянуться. Это может привести к разрыву ГКС-мышцы, что вызовет внутреннее кровотечение и гематому внутри мышцы. В течение некоторого времени внутри мышцы развивается рубцовая ткань, которая уплотняет мышцу – и, в конечном итоге, приводит к формированию аномального положения шеи.
- Пороки развития плода. Во время внутриутробного развития плода, на фоне аномальных положений в матке (поперечное, косое), шея малыша может изгибаться, а затем продолжает развиваться уже таким аномальным образом. Это приводит к сокращению длины мышцы, в результате чего новорожденный рождается с кривошеей.
- Врожденные мышечные или скелетные аномалии. Например, синдром Клиппеля-Фейля, при котором кости шеи срастаются, вызывая аномальный наклон головы.
- Внутриутробные проблемы. Чрезмерный тонус стенок матки может привести к чрезмерному сдавлению головки и шеи плода. Это приведет к сокращению длины шейных мышц.
- Повреждение плода. Физическое воздействие на матку может повлиять на развитие плода. Это может привести к аномальному сокращению мышц шеи, и ребенок родится с наклоном головы.
- Травматичные роды. Ребенок, застрявший в родовом канале во время нормальных родов, должен быть извлечен с помощью щипцов или вакуума. Силовое извлечение во время трудных родов может вызвать травму мышц на шее. Неправильное использование вспомогательного оборудования для родов также может привести к повреждению тканей — и, как следствие, к кривошее.
Приобретенная кривошея возникает, как побочный эффект различных аномалий в состоянии здоровья, и не наблюдается при рождении ребенка.
- Глазная кривошея. У детей с косоглазием могут возникнуть проблемы с восприятием глубины и расстояния. Чтобы компенсировать недостаток зрения, младенец наклоняет голову, чтобы правильно видеть картинки. Ребенок, постоянно принимая вынужденную позу в ходе роста мышцы, за счет неправильного положения шеи, тормозит в развитии мало работающую ГКС-мышцу на одной стороне – что, в конечном итоге, приведет к глазной кривошее. Это состояние также называется глазной аномальной установкой головы. Глазная кривошея не относится к мышечно-скелетной причине кривошеи, так как ребенок родился со здоровой мышцей. Такая кривошея может быть легкой. Проблема кроется в глазных мышцах, которые отвечают за движение глазных яблок.
- Синдром Сандифера. У детей, страдающих тяжелой гастро-эзофагеальной рефлюксной болезнью (ГЭРБ), может развиться редкое детское заболевание, называемое синдромом Сандифера. В этом состоянии у ребенка наблюдается ненормальное сокращение мышц шеи и приступы рефлюкса. Это приводит к тому, что младенец принимает неудобные положения тела в силу мышечной дистонии (высокий тонус в одних группах мышц, и низкий – в других). Аномалии позы приводят к наклону головы и удержанию шеи в положении, подобном кривошее. Эти аномалии вызваны спазмами сосцевидной мышцы из-за неверных нервных импульсов. Приобретенная кривошея из-за рефлюкса встречается крайне редко.
- Доброкачественная пароксизмальная кривошея. При ней ребенок наклоняет голову на одну сторону регулярно, в течение длительного времени, от нескольких часов — до нескольких дней подряд. Наклон происходит в любом случайном направлении, и ребенок возвращается к нормальному состоянию после эпизода кривошеи. Повторение таких движений распространено, и это состояние сопровождается другими симптомами – такими, как дезориентация, сонливость и раздражительность. Проблема проявляется в течение первых 12 месяцев жизни ребенка, и обычно исчезает после пятилетнего возраста. Причины доброкачественной пароксизмальной кривошеи неизвестны, но считается, что она вызвана проблемами нервной системы, а не мышц. Расстройство связано с генетической мутацией, которая играет роль в других неврологических проблемах — мигрень и головокружение.
- Синдром Гризеля. Инфекция носоглотки может вызвать воспаление мышц шеи, что приводит к подвывиху (дислокации) атланто-аксиального сустава. Сустав лежит между атлантом (первый шейный позвонок) и аксисом (второй шейный позвонок), то есть, это аномалии сочленения между двумя первыми шейными позвонками в области шеи. Дислокация может произойти даже из-за инфекции в области миндалин, аденоидов и среднего уха. Синдром также появляется после хирургических процедур – таких, как тонзилэктомия или аденоидэктомия. Вывихнутые суставы постоянно изгибаются в одном направлении, что приводит к классическому наклону головы, кривошее. Инфекции уха, носа, горла (ЛОР-патологии) считаются основной причиной проблемы. Синдром Гризеля может развиться в любом возрасте, но наиболее часто встречается у подростков.
- Аномалии позвоночного столба. Деформации позвоночника могут вызвать изменение вертикальной оси тела и спинномозгового канала, который необходим для защиты расположенного внутри спинного мозга. Изменение положения позвонков нарушает работу двигательных нервов, заставляя мышцы дергаться (сокращаться). Сокращение ГКС-мышцы, в конечном итоге, приводит к кривошее. Скелетные нарушения – такие, как сколиоз — часто являются причиной аномалий позвоночника.
- Кивательный спазм. Это состояние отображает три проблемы одновременно: кивание головой, кривошея и нистагм, что является быстрым непроизвольным движением глаз. Причины кивательного спазма неизвестны, но состояние связано с дефицитом железа и витамина D, а также с нарушениями работы мозга. Многие эксперты считают проблему офтальмологической, поскольку быстрое движение глаз сильно влияет на зрение. Пациент компенсирует это кивками и наклоном головы.
- Инфекция и травмы. Дети могут страдать кривошеей даже в силу общих причин – таких, как прямая травма шеи. Инфекции шейного отдела позвоночника и окружающих его тканей также могут, в конечном итоге, вызвать наклон головы.
Скрученная, повернутая в сторону шея является основным симптомом кривошеи.
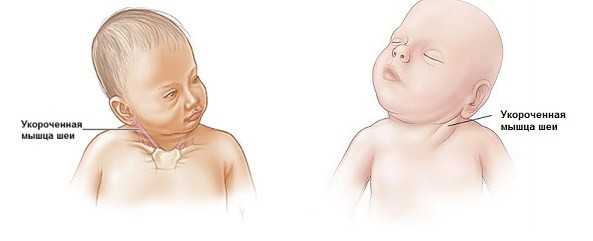
Симптомы кривошеи у младенцев – как точно определить патологию
Наклоненная, скрученная, повернутая в сторону шея – это классический симптом кривошеи.
- Младенец поворачивает голову только в одну сторону. Например, если ребенок страдает правосторонней кривошеей (голова наклонена вправо), то он всегда будет перемещать голову влево. Если ребенок должен смотреть направо, то он поворачивает все свое тело вправо, одну голову он повернуть не может.
- Младенец предпочитает кормление одной грудью. Поскольку кривошея приводит к ограниченному диапазону движений головой, то у ребенка могут возникнуть проблемы с прикладыванием к груди. Он может суетиться во время кормления одной грудью, но ему будет удобно, и он будет довольным при кормлении другой.
- На шее имеется утолщение мышцы. Наличие комка указывает на наличие рубцовой ткани. Если у ребенка была травма грудино-ключично-сосцевидной мышцы, то родители могут заметить небольшую шишку на мышце.
Родители могут следить за этими симптомами, которые хорошо видны.
Оно поможет избежать инвалидности.
Читайте также: