
Генетическое заболевание мышц и суставов
Генетические заболевания обусловлены патологическими нарушениями строения генома. "Дефектный" ген может быть получен от одного из родителей и проявиться как на 100%, так и на 10%. А вот болезни с наследственной предрасположенностью значительно отличаются от генетических. Если последние излечить невозможно, то заболевания, к которым человек имеет наследственную предрасположенность, возможно нивелировать рациональным питанием, здоровым образом жизни и профилактическими мерами.
Пять генетический заболеваний позвоночника и костей
Такие болезни напрямую связанны с нарушениями генома и проявляются в виде дефектов развития скелета человека. Генетические заболевания обусловлены нерациональным формообразованием ткани или нарушениями роста. Подобные болезни носят в медицине общие название - дисплазии.
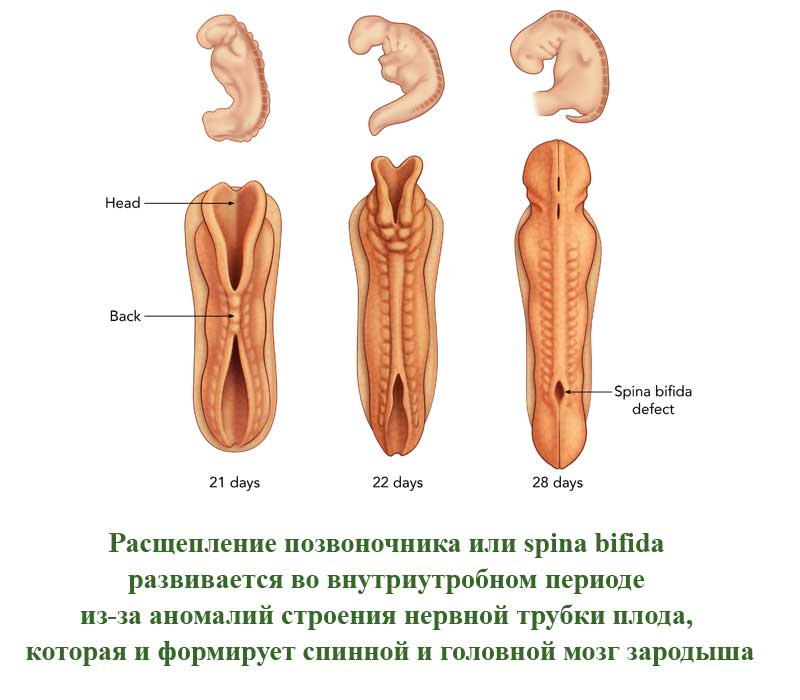
Это порок развития позвоночного столба, которое проявляется в виде недоразвитых позвонков. Такие позвонки не сомкнуты, через щель может быть виден спинной мозг. Заболевание развивается во внутриутробном периоде из-за аномалий строения нервной трубки плода, которая и формирует спинной и головной мозг зародыша. Расщепление позвоночного столба может проявляться и в закрытом виде, когда спинной мозг не виден снаружи.
В легких случаях заболевание могут обнаружить лишь при рентгеновском обследовании. А вот при самых серьезных формах болезни у ребенка могут сразу же образовываться свищи в полости позвоночника. Очень часто заболевание в тяжелых формах сопровождается параличом нижней части тела.
В более, чем 80% случаев, расщепление позвоночника сопровождается гидроцефалией спинного мозга и пороками развития головного мозга, а также - черепа.
По американской статистике, заболевание встречается у одного пациента из 1500. Российская статистика приводит следующие данные - 3 случая на 10000 человек. Однако, многие случаи расщепления позвоночника на территории СНГ остаются нераспознанными у новорожденных из-за легкой формы болезни.
Болезнь часто именуют остеопетрозом. Может протекать в двух формах:
- замедленной;
- злокачественной.
Генетическое заболевание встречается с частотой в 1 случай на 20000 пациентов. Для остеопетроза характерны такие симптомы:
- повышенная ломкость костей;
- увеличение плотности костной ткани;
- уменьшение размеров костномозговых лакун;
- нарушение гемопоэза;
- уменьшение массы костного мозга.
Генерализированный остеоклероз проявляется в достаточно раннем возрасте в виде разных беспорядочных слоев клеток костной ткани, увеличения общей массы костей и замедленном росте скелета.
При злокачественном течении болезни часто возникают внезапные переломы костей, развивается геморрагичекий синдром, жировая дистрофия органов, нарушается дентиногеез. Характерен очень небольшой рост.
В случае замедленного остеопетреза болезнь может быть выявлена лишь в 50% и протекать абсолютно бессимптомно. Выявляют заболевание случайно во время рентгена. В некоторых случаях может наблюдаться симптоматика синдрома "Кость внутри кости".
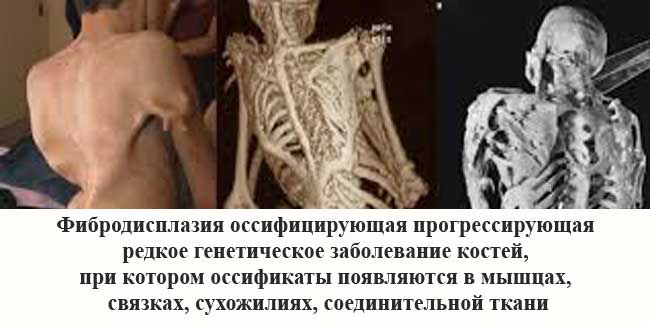
ФОП - это генетическое и очень редкое заболевание костей. При такой болезни организм начинает формировать новую костную массу в виде оссификатов в ненадлежащих местах тела, а именно внутри:
- соединительных тканей;
- связок;
- мышц;
- сухожилий.
К образованию оссификатов в организме может привести абсолютно любая травма: порез, операция, ушиб, внутримышечная инъекция или перелом. Поэтому образования такого типа удалять нельзя - на их месте костная ткань разрастется еще больше. По физиологическим признакам оссификаты совершенно не отличаются от здоровых костей.
Проблема лишь в неправильном расположении образования костной ткани. Возникает ФОП из-за мутаций гена ACVR1/ALK2. Данный ген кодирует рецептов костного морфогенетического белка. Носителем гена быть невозможно, его наличие в теле всегда вызывает развитие фибродисплазии оссифицирующей. Передается заболевание по наследству и на данный момент является неизлечимым.
Такие заболевания характеризуются чрезмерным развитием костной массы. Носят общее название - остеохондродисплазии. Гиперостозы возникают из-за генетических нарушений и патологий остеобластов и остеокластов. Наиболее часто встречаются такие формы остеохондродисплазий:
- Болезнь Лери или мелореостоз;
- пикнодизостоз.
Мелореостоз чаще всего поражает мужчин, может развиться в любом возрасте. Характеризуется болезнь избыточным образованием эндостальной или периостальной кости. Процесс может происходит в двух зонах одновременно. Зарождается болезнь Лери с поражения нижних конечностей. Процесс может переходить на все суставы, отдельные кости таза, позвоночный столб, ребра и даже череп. Все пораженные кости довольно слабо изменены и деформированы, кортикальный стой утолщен, а костномозговая полость сужена неравномерно.
Мелореостоз может протекать совершенно бессимптомно продолжительное время, однако, при значительном уменьшении габаритов костномозговых лакун развивается болевой синдром в пораженной конечности. Нога при этом может укорачиваться или увеличиваться, развивается анкилоз сустав, нарушается гемопоэз.
Пикнодизостоз проявляется в виде карликовости и остеоскрероза. В основе заболевания лежит неравномерное, чрезмерное и очаговое периостальное развитие компактной кости. Развивается явная деформация скелета в виде:
- сколиоза;
- кифоза;
- гипоплазии ключиц;
- укорочении пальцевых фаланг;
- уменьшении длины предплечий.
В молочных зубах ребенка быстро развивается кариес, склеры глаз приобретают характерных болезни голубой оттенок. На продолжительности жизни пикнодизостоз не сказывается.
Болезнь Брутона
Болезнь Брутона (первичная гипо- или агаммаглобулинемия взрослых) — наследственное заболевание семейного характера, сцепленное с Х-хромосомой, однако нередко также рецессивное наследование. Часто уже в раннем детстве отмечается склонность к повторным бактериальным инфекциям различной локализации (легкие, придаточные пазухи носа, кожа). К типичным признакам относятся диарея, а также увеличение периферических лимфатических узлов и селезенки. Возможны системные ревматические проявления по типу диффузных болезней соединительной ткани. Суставной синдром характеризуется эпизодической мигрирующей полиартралгией либо острым, подострым, но чаще хроническим моно- или асимметричным олигоартритом крупных суставов. В синовиальной жидкости признаки воспаления слабо выражены или отсутствуют вовсе. Даже при длительном течении артрит не приводит к рентгенологическим изменениям пораженных суставов. В анализах крови СОЭ в норме, острофазовые показатели не изменены, однако снижены уровень гамма-глобулинов (или они отсутствуют), иммуноглобулинов (тотально или избирательно), отсутствуют изогемагглютинины. В костном мозге выявляется отсутствие или снижение содержания плазмоцитов, а в биоптате лимфатического узла — сужение кортикального слоя, первичные фолликулы в нем редкие и малоразвитые.
Болезнь Волкова
Болезнь Гоше
Болезнь Гоше — относительно редкое заболевание, распространенное преимущественно среди евреев-ашкенази и обусловленное дефицитом лизосомальной бета-глюкоцереброзидазы. В результате происходит массивное накопление глюкоцереброзида в селезенке, печени, легких, костном мозге и других органах и тканях с образованием характерных гигантских клеток, содержащих вакуоли глюкоцереброзида (клетки Гоше). У больных, как правило, увеличены печень и селезенка, а иногда и периферические лимфатические узлы. Кроме того, выявляется очаговая кожная пигментация охряного или коричневого цвета, а у некоторых больных — желтые пятна на склерах. У детей наблюдается поражение нервной системы. В анализах крови отмечаются анемия, лейкопения и тромбоцитопения, которая может приводить к геморрагическому синдрому с носовыми кровотечениями и подкожными кровоизлияниями. В ряде случаев обнаруживается моноклоновая гаммапатия. Нередко развиваются костные повреждения (чаще всего деструкция шейки, головки или верхней трети бедра), приводящие к болям, деформации костей, патологическим переломам. Рентгенологическое исследование обнаруживает увеличение объема кости, чередование в ней участков уплотнения и разрежения либо диффузную декальцификацию костей с явлениями спонгиоза, истончение кортикального слоя, кистозные и остеосклеротические изменения. Могут возникать боли и припухлость крупных суставов, чаще нижних конечностей. Иногда поражается также поясничный отдел позвоночника, что приводит к сплющиванию (компрессии) тел позвонков. Для детской формы болезни Гоше типична быстрая смерть, при хроническом течении — медленное прогрессиро-вание. Диагноз основывается на выявлении в стернальном пунктате гигантских клеток Гоше.
Болезнь Мухи—Хаберманна
Болезнь Мухи—Хаберманна (Mucha—Habermann) — довольно распространенное, но редко распознаваемое кожное заболевание, встречающееся у детей. Характерно появление оспоподобных везикул на коже туловища, бедер, предплечий. Эти элементы могут эволюционировать в геморрагии, некроти-зироваться и разрешаются обычно с образованием корочек. Как правило, кожные поражения протекают изолированно, но иногда сопровождаются субфебрильной лихорадкой, а в ряде случаев — артритом (в том числе по типу ревматоидного артрита), склеродермией, интерстициальным пневмонитом, повышенной чувствительностью к укусу пчел. Предполагают возможную связь заболевания с реактивацией инфекции, вызванной вирусом Эпштейна—Барр.
Болезнь Тиманна
Болезнь Тиманна (Thiemann) — наследственное заболевание (аутосомно-доминантный тип наследования), встречающееся у юношей в начальном периоде полового созревания и проявляющееся множественной остеохондропа-тией (асептическим остеонекрозом) фаланг пальцев кистей, а иногда и стоп. Поражаются эпифизы средних фаланг указательного, среднего и безымянного пальцев (кроме большого пальца), обычно вовлекаются одновременно 2— 3 пальца обеих кистей, но не симметрично. Выявляется веретенообразная припухлость в области проксимальных, а иногда и дистальных межфаланго-вых суставов. Могут поражаться также межфаланговый сустав I пальца стоп и I тарзо-метатарзальный сустав. При рентгенологическом исследовании отмечается уменьшение, а нередко резорбция пораженных эпифизов, соответствующая фаланга укорачивается. Возможно спонтанное выздоровление, однако чаще остаются явления выраженного вторичного остеоартроза.
Болезнь Фабри
Болезнь Фабри (Fabry) — врожденное заболевание, характеризующееся наследственным дефицитом фермента альфа-Г4-галактозидазы, приводящим к накоплению гликолипидов (церамида) в цитоплазме и лизосомах клеток различных органов и тканей. Заболевание начинается с детства. Изменения кожи проявляются ее утолщением и ангиоэктазиями, поражение глаз — помутнением роговицы. При поражении синовиальной оболочки суставов происходит утолщение капсулы суставов и сухожильных влагалищ, отмечаются жгучие боли в пальцах кистей и стоп, голенях, предплечьях при повышении внешней температуры и ослабление болей при охлаждении конечностей. Периодически наблюдается отечность голеностопных и коленных суставов на фоне немотивированного субфебрилитета. Таким образом, клиническая картина часто напоминает воспалительное ревматическое заболевание. Рентгенологическое исследование выявляет в костях кистей множественные энтезопатические кальцификаты, а также мелкие интра- и экстраартикулярные эрозии.
Болезнь Файербанка
Болезнь Файербанка (множественная эпифизарная дисплазия) — наследственное заболевание, характеризующееся нарушением развития эпифизов, главным образом длинных трубчатых костей. Заболевание проявляется в детском и юношеском возрасте. У больных отмечаются низкий рост, нарушение походки, контрактуры и деформации преимущественно суставов нижних конечностей, широкие кисти с утолщенными и укороченными пальцами, вывихи и подвывихи в коленном и локтевом суставах, деформация стоп за счет укорочения пальцев. При рентгенологическом исследовании выявляются уменьшение размеров поперечника эпифизов, их деформация, уплощение, нередко — дольчатое строение (фрагментация). Указанные изменения носят симметричный характер и наиболее выражены главным образом в тазобедренных, коленных, плечевых и локтевых суставах.
Мукополисахаридозы
Мукополисахаридозы — группа наследственных заболеваний (аутосомно-рецессивный тип наследования), в основе которых врожденный дефект энзимов, принимающих участие в обмене глюкозаминогликанов — основных компонентов органического матрикса соединительной ткани. Отмечаются аномальная продукция, чрезмерное накопление и выделение одного или нескольких определенных мукополисахаридов, вследствие чего развиваются патологические изменения в хряще (множественный дизостоз), фасциях, периосте, сухожилиях, сердечных клапанах (сморщиваются в результате рубцовых процессов), кровеносных сосудах, черепно-мозговых оболочках, роговице, печени, селезенке (см. синдромы Марото-Лами, Моркио, Пфаундлера-Гурлера, Шийана).
Синдром Джеккейя
Синдром Джеккейя (Giaccai) — редкое семейное заболевание с аутосомно-рецессивным типом наследования, наблюдающееся в районах с высокой частотой близкородственных браков и характеризующееся прогрессирующим симметричным лизисом дистальных частей конечностей (акроостеолизом) с их последующей деформацией. Болезнь начинается в юношеском, реже — в детском возрасте и клинически похожа на остеомиелит и саркому. У больных наблюдается симметричный лизис костей дистальных отделов конечностей и последующая их деформация. На подошвенной поверхности стоп над участком рассасывания костей образуются очаги безболезненной припухлости, чаще без кожной гипертермии. Через некоторое время на этих участках появляются длительно не заживающие кожные дефекты (язвы), через которые происходит спонтанное отторжение мелких костных секвестров. После этого язвенные дефекты заживают, но могут наблюдаться и торпидно текущие.
Течение заболевания рецидивирующее с ремиссиями от нескольких недель до 3—4 лет. В итоге происходит выраженная деформация стоп с ампутацией отдельных ногтевых фаланг, деформацией и укорочением пальцев. Кроме того, отмечаются трофические изменения ногтей, гипотрофия мышц голеней и омозолелость подошвенных поверхностей, гипостезия кистей и стоп с сохранением тактильной чувствительности. Рентгенологически выявляются остеолиз фаланг и дистальных отделов плюсневых костей, очаги их деструкции без четкой периостальной реакции.
Синдром Клиппела—Тренонея
Синдром Клиппела—Тренонея — врожденная сосудистая аномалия в виде обширных варикозных расширений в области конечностей (в том числе кистей и пальцев) и верхнего плечевого пояса. Постепенно развивается гипертрофия мягких тканей конечностей, отмечается тенденция к изъязвлению кожных покровов. Происходит деформация костей (искривление, утолщение) и суставов. Возможно образование анкилозов. В области пораженных суставов могут прощупываться и рентгенологически определяться флеболиты.
Синдром Кускоквим
Синдром Кускоквим (по названию реки в штате Аляска, США, в районе дельты которой впервые были выявлены больные с данным синдромом) — комплекс наследственных аномалий (аутосомно-рецессивный тип наследования), включающий множественные контрактуры крупных суставов (преимущественно коленных и локтевых), косолапость и косорукость.
Синдром Лери
Синдром Лери — наследственное заболевание костей, характеризующееся поражением обычно одной из верхних или нижних конечностей и проявляющееся болями, распространяющимися вниз по конечности, а позже — ограничением подвижности в суставах на пораженной стороне. Часто на соответствующей стороне наблюдаются также склеродермия, атрофия и кальциноз мягких тканей. Рентгенологически обнаруживается эндостальный и частично пе-риостальный остеосклероз, возможно выявление продольных полос кальциноза в мягких тканях пораженной конечности.
Синдром Лутца—Жансельма
Синдром Лутца—Жансельма (Lutz—Jeanselme) — гиперэозинофилия в сочетании с образованием околосуставных узелков. Постепенно появляются различной величины и консистенции безболезненные подкожные узелки, локализующиеся преимущественно над костными выступами на поверхности суставов, чаще на локтях, над мелкими суставами пальцев, над областью тазобедренных суставов. В области мелких суставов кожа над узелками нормального вида, а над крупными суставами — натянутая, синюшная. Характерны артрал-гия, перемежающаяся лихорадка, слабость, кахексия. Возможно развитие неэрозивного полиартрита с вовлечением периартикулярных тканей. В крови и синовиальном выпоте определяется высокая эозинофилия. В анамнезе у больных различные аллергические заболевания. Болезнь тянется годами, обострения чередуются с частыми периодами ремиссии.
Генетическое заболевание, которое приводит к атрофии мышц спины. Существует несколько ее видов (типов), отличающихся протеканием, скоростью развития и тяжестью поражения. Заболевание не поддается лечению и опасно своими осложнениями.

Безболезненная, уникальная методика доктора Бобыря
Дешевле, чем мануальная терапия
Мягко, приятно, нас не боятся дети
Только с 20 по 30 июня! Записывайтесь сейчас!

Во всем мире данные типы поражений называются «спинальные мышечные атрофии, или SMA. Хотя диагноз такого рода ставится редко, это одна из наиболее распространенных генетических патологий. Переносится она геном SNN, выделенным в 1995 году. Количество рожденных с этим заболеванием различается в различных странах, в среднем оно составляет 6 новорожденных на 1000. Большинство из них не доживает до 2-летнего возраста.
Различают заболевания по времени первого проявления, скорости протекания и тяжести. Часть признаков заболевания диагностируется сразу же при рождении, но некоторые проявляются только при достижении зрелого возраста. В последнем случае говорят о "мягком" течении заболевания. И главный метод борьбы с последствиями болезни - лечение осанки.
Часто различают следующие группы заболевания, различающиеся по времени проявления или течения.
1. Болезнь Вердинга-Гофмана, или первый тип SMA. Это наиболее тяжелая форма, выявляется у младенцев в возрасте до полугода.
2. Промежуточная форма (второй тип), проявляется в возрасте от 6 месяцев до 1,5 лет.
3. Третий тип SMA называется болезнью Кугельберга-Веландер. Он проявляется после 1,5 лет или даже позже.
4. Синдром Кеннеди, или взрослая форма SMA, проявляется в возрасте старше 35 лет.
При этом каждая из форм заболевания отличается проявлениями от других типов.
Болезнь Верднига-Гоффмана - наиболее выраженная и тяжелая форма. Дети с этой болезнью живут не дольше 2 лет, так как у них не только развивается общая слабость, но нередко также ослаблено глотание и дыхание. Умственное и общее физическое развитие при этом не страдает. Диагностировать болезнь иногда удается еще на стадии беременности - движения плода очень слабые.
У детей, пораженных болезнью, атрофируются мышцы, они не способны самостоятельно сидеть. Наблюдается деформация скелета, у детей отсутствует сухожильный рефлекс, нет подвижности в суставах. Развиваются сопутствующие расстройства дыхания и легочные инфекции. Подтверждением заболевания служит генетическая экспертиза.
Промежуточная форма (Тип 2). Слабость в ручках и ножках развивается после 6 месяцев, но до достижения возраста в 1,5 года. Однако развивается болезнь не настолько быстро и не имеет настолько тяжелых последствий. Руки и ноги при такой болезни и в дальнейшем остаются слабыми, но для выполнения повседневных базовых занятий этого обычно вполне достаточно. Свободно ходить такой ребенок может с трудом, он должен пользоваться ходунками или специальными фиксаторами.
Сильно страдает при таком заболевании функция дыхания. Поэтому необходимо сразу же лечить любые заболевания органов дыхания, предотвращая их в самом начале. Также опасным является и сколиоз, или искривление позвоночника, вызванное слабостью мышц. Это тоже может мешать нормальному дыханию.
Хотя лечения заболевания не существует, при заботе со стороны родителей и постоянном лечении возникающих сопутствующих заболеваний срок жизни - 16 или 18 лет.
Если признаки заболевания появляются после 18 месяцев, то это болезнь Кугельберга-Веландер. Диагноз ставится, когда ребенок начинает ходить. Прогноз течения заболевания более благоприятный. И хотя возможность самостоятельного передвижения больные с таким поражением теряют в юношеском возрасте, люди с таким диагнозом живут до 35-40 лет, если обеспечивается соответствующий уход и лечение сопутствующих заболеваний.
Главное внимание также уделяется защите органов дыхания и борьбе с последствиями искривления позвоночника. Иногда пациенты могут передвигаться самостоятельно, опираясь на трость. Однако чаще им необходимо пользоваться инвалидной коляской.
В случае болезни Кугельберга-Веландер симптомы прогрессируют значительно медленнее. Первыми начинают страдать ноги, затем постепенно начинают хуже слушаться и руки. Время от времени заметны подергивания икроножных мышц, мышц плечевого пояса. При этом больной длительное время может самостоятельно обслуживать свои функции, способен к активной деятельности.
Здоровые суставы — это роскошь, значение которой трудно оценить тому, кто никогда не испытывал боли при ходьбе и не ощущал затруднения при попытке поднять руку или ногу, развернуться или присесть. Между тем миллионы людей по всему свету ежегодно обращаются за помощью к врачам-ортопедам с подобными жалобами.
Эпидемиология заболеваний суставов
Болезни суставов классифицируются по характеру патологического процесса.
Воспалительные и инфекционные болезни суставов
В данном случае причиной недуга служит воспалительная реакция в ответ на инфекционный, аутоиммунный или аллергический процесс. Болезнь дает о себе знать болью и припухлостью в суставе. Симптомы за несколько часов способны достичь максимальной выраженности и в дальнейшем отступить на неопределенный срок. Но за периодом мнимого благополучия скрывается скрытое развитие патологического процесса.
Некоторые артриты проявляются в первую очередь не болью, а утренней скованностью, лихорадкой, сыпью на коже над суставом или проявлениями первичного заболевания, осложненного поражением опорно-двигательного аппарата (например, расстройствами мочеполовой системы — при болезни Рейтера и гонорейном артрите).
Этапы воспалительной патологии сустава можно разграничить благодаря рентгенологическому обследованию. Так, в начале болезни значимых отклонений на снимках не обнаруживается, при второй степени артрита появляются признаки разрушения костной и хрящевой ткани. При третьей степени врач увидит деформацию сустава — к этому моменту больной ощущает выраженные ограничения подвижности. Наконец, четвертая степень артрита сопровождается тотальными изменениями в суставе: помочь больному смогут только хирурги.
К этой группе патологий, наряду с ревматоидным артритом (при котором иммунная система организма разрушает суставы), относится инфекционно-аллергический артрит, болезнь Бехтерева, болезнь Гоффа, подагра, псориатический артрит и т. д.
Обратите внимание
Основная профилактика артритов — предотвращение инфекций путем вакцинации и других мер защиты от болезней, способных осложниться бактериальным заражением сустава (гонореи, скарлатины, ангины и т.д.)
Дегенеративные поражения
Эти болезни, как правило, проявляются у пожилых людей: они связаны с изнашиванием сустава, что приводит к разрушению хрящей, отвечающих за амортизацию при работе. Еще один фактор развития дегенеративных заболеваний — остеопороз, при котором снижается плотность костной ткани.
Дегенеративные заболевания проявляются постепенно: первым признаком будет непродолжительная боль после утомительного дня, проведенного на ногах. С течением времени болезненность в суставе перестанет стихать после отдыха, а движения в суставе станут ограниченными. Иногда в пораженной области возникает отек, а сустав ноет при смене погоды и по ночам.
Тяжесть артрозов определяется при осмотре и рентгенографии. В начале процесса на снимках заметно лишь небольшое сокращение высоты щели между костями вследствие истончения хрящевой ткани. При второй степени врач зафиксирует уменьшение полости сустава на треть от нормы и появление костных выростов или участков отмершего хряща. Наконец, 3 стадия артроза характеризуется глубокими, необратимыми деформациями сочленения, вплоть до анкилоза — сращения костей.
Самым частой причиной инвалидности среди всех болезней суставов является деформирующий остеоартроз, затрагивающий преимущественно тазобедренный и коленный суставы. Также источником проблемы может оказаться межпозвоночный остеохондроз — типичное заболевание офисных работников.
Обратите внимание
Эксперты отмечают, что отличной профилактикой возрастных изменений суставов может стать йога — комплекс упражнений, укрепляющих связки и мышцы посредством статических нагрузок. Недавно ученые выяснили, что ежедневные 12-минутные занятия йогой на протяжении 10-ти лет способствуют увеличению плотности костей бедра и позвоночника, что исключает остеопороз и артроз, предотвращая переломы в пожилом возрасте.
Врожденные патологии суставов
Врожденные болезни суставов обращают на себя внимание с самых первых дней жизни малыша: от лечебных мер зависят возможные последствия порока развития для здоровья ребенка. Так, у младенцев нередко выявляется врожденный вывих бедра, который часто сочетается с дисплазией тазобедренного сустава. Будучи оставленным без внимания, этот недуг приведет к проблемам с походкой и формированием осанки. Однако своевременное вмешательство ортопеда позволит скорректировать врожденный вывих бедра консервативно, без операции.
Еще одна часто встречающаяся врожденная патология суставов — синдром Марфана, включающий в себя комплекс нарушения развития внутренних органов. Пациенты с синдромом Марфана имеют крайне подвижные суставы, нарушение осанки и килевидную грудную клетку. Это — высокие, худые, болезненные люди, которые, как правило, попадают в травмпункты с вывихами и переломами. При соблюдении рекомендаций врача они могут жить долго, не испытывая тяжелых проблем со здоровьем.
Болезни околосуставных тканей
Лечение заболеваний суставов в китайской медицине направлено на рассеивание ветра, холода, жары, а также преобразование сырости и восстановление течения Ци и крови. В этом помогает иглоукалывание, моксотерапия (прогревание полынными сигарами) и точечный массаж.
Не забывайте, что важно с вниманием относиться к любым симптомам — боли, скованности, отеку, появлению сыпи или хрусту, слышимому при движениях. Подавляющее число заболеваний суставов успешно поддается излечению на начальных стадиях, однако в запущенном состоянии болезни способны нарушать жизненные и профессиональные планы, требуя дорогостоящего оперативного вмешательства и сложных реабилитационных мероприятий.
Лицензия на осуществление медицинской деятельности от 30 декабря 2008 года № ЛО-77-01-000911 выдана Департаментом здравоохранения города Москвы.
Миопатии, или болезни мышц, — группа заболеваний, объединяющих различные нарушения в строении и метаболизме мышечной ткани и сопровождающихся снижением силы и ограничением двигательной активности. Они проявляются мышечной слабостью, развитием мышечной атрофии, снижением сухожильных рефлексов и мышечного тонуса.
Виды миопатий
Болезни мышц относятся к группе нервно-мышечных заболеваний. Они делятся на первичные и вторичные.
Первичные миопатии обусловлены генетически детерминированными нарушениями в функционировании митохондрий и ионных каналов миофибрилл, а также сбоями в продуцировании белков или ферментов, которые регулируют метаболизм мышечной ткани. Наследование дефектного гена, обуславливающего болезни мышц, может происходить по доминантному, рецессивному и сцепленному с Х-хромосомой типу. Зачастую внешние факторы выступают в роли пусковых механизмов развития болезни.
Приобретенные болезни мышц могут возникать на фоне эндокринных патологий, хронических интоксикаций, авитаминоза, мальабсорбции, тяжелых хронических заболеваний. В эту группу входят аутоиммунные миастении, посттравматические и метаболические миопатии, краш-синдром и т. д.
Наследственные болезни мышц
Большинство наследственных нервно-мышечных заболеваний наследуются аутосомно-рецессивно и характеризуются высоким риском передачи по наследству. Такие болезни подразделяют:
- на первично-мышечные заболевания (прогрессирующие мышечные дистрофии, врожденные структурные миопатии, лице-плече-лопаточная дистрофия, дистальные мышечные дистрофии и т. д.);
- метаболические миопатии (связанные с дефицитом фосфорилазы, кислой мальтазы, фосфофруктокиназы, карнитина и т. д.);
- болезни мотонейрона (спинальная мышечная атрофия, боковой амиотрофический склероз);
- миотонии и периодический паралич;
- заболевания периферических нервов;
- болезни нервно-мышечных синапсов и др.
Диагностика болезней мышц
Для установки диагноза используются электрофизиологические методы обследования: электромиография и электронейрография. Они помогают дифференцировать болезни мышц от миелита, миелопатий, опухолей спинного мозга, нарушений спинномозгового кровообращения.
Биохимический анализ крови при миопатиях показывает повышенный уровень альдолазы, АЛТ, КФК, АСТ, ЛДГ. Биохимический анализ мочи может обнаружить высокий уровень креатинина.
В целях диагностики болезней мышц проводят их биопсию. Морфологическое исследование образцов может обнаружить беспорядочно разбросанные атрофированные миофибриллы среди сохранных и гипертрофированных мышечных волокон, участки замещения мышечной ткани на жировую или соединительную.
МРТ мышц позволяет визуализировать волокна, сухожилия, отдельные пучки. Исследование помогает обнаружить травмы, разрывы, посттравматические повреждения, образования.
Диагностика наследственных болезней мышц
Генетические исследования позволяют обнаружить мутации в известных генах, ассоциированных с миопатиями. Молекулярно-генетический анализ предоставляет возможность избежать дальнейших диагностических исследований и спрогнозировать течение заболевания. Благодаря генетической диагностике можно установить вероятность передачи болезней мышц по наследству.
Диагностика проводится с помощью таких методов, как:
- хромосомный микроматричный анализ (изучение всей структуры генома человека в одном исследовании позволяет исследовать последовательность генетического материала во всех хромосомах, обнаружить его избыток или недостаток);
- экзомное секвенирование нового поколения (заключается в анализе кодирующих последовательностей всех известных генов или специально отобранных генов, назначается при спорных клинических случаях).
| Наименование | Срок выполнения, дни | Цена исследования, руб |
| Панель "Нейродегенеративные заболевания" | 90 | 35000 |
| Панель "Наследственные нарушения обмена веществ" | 90 | 35000 |
| Панель "Наследственные эпилепсии" | 90 | 35000 |
| Панель "Нервно-мышечные заболевания" | 90 | 35000 |
| Панель "Умственная отсталость и расстройства аутистического спектра" | 90 | 35000 |
По результатам диагностики болезней мышц рекомендуется получить консультацию невролога и генетика.
Читайте также: