
Полимиозит с системной склеродермией
Дерматомиозит и полимиозит – это заболевания, характеризующиеся системным негнойным воспалением соединительной ткани, поперечно-полосатой и гладкой мускулатуры с нарушением её двигательной функции.
- При дерматомиозите (болезнь Вагнера, болезнь Вагнера-Унферрихта-Хеппа) поражение мышц сочетается с поражением кожи в виде эритемы и отека на открытых участках тела.
- При полимиозите развивается только поражение мышц, а кожный синдром отсутствует.
Заболеваемость составляет примерно 1:200000 в год. Девочки и женщины болеют в 2 раза чаще.
Этиология и патогенез
Причина заболевания неизвестна. Существует мнение, что в его развитии имеют значение вирусная инфекция и аутоиммунные реакции. В крови больных дерматомиозитом детей обнаружены антитела к пикорнавирусам, а у больных полимиозитом взрослых к Toxoplazma gondii. У больных гриппом и инфекцией, вызванной вирусом Коксаки, может развиться легкий миозит. Но до настоящего времени вирусологические и серологические исследования и отсутствие эффекта от антибактериальной терапии токсоплазмоза не подтверждают вирусную природу этих заболеваний. Но, возможно, измененные вирусы содержат антигены, напоминающие собственные антигены организма и вызывающие образование аутоантител к цитоплазматическим белкам (антигену J0-1).
Аутоиммунная природа заболеваний подтверждается обнаружением аутоантител к ядерным или цитоплазматическим антигенам, культурам эпителиальных клеток человека (Нер-2), синтетазам транспортной РНК (например, к гистидил-тРНК-синтетазе) и внутрисосудистых отложений иммунных комплексов, повреждающих эндотелиальные клетки. Иммуногистохимические исследования мышечной ткани при полимиозите выявляют воспалительные инфильтраты, содержащие активированные цитотоксические лимфоциты СD8 наряду с лимфоцитами СD4 и макрофагами. При дерматомиозите в биоптатах мышц обнаруживают отложения иммуноглобулинов и компонентов комплемента С5b–С9, а вокруг мелких сосудов накапливаются СD4 + -клетки и β-лимфоциты. Развивается поражение сосудов, опосредованное антителами и приводящее к образованию очагов некроза в мышечных волокнах, их дегенерации и потере поперечной исчерченности.
Регенерация скелетных мышц проявляется увеличением числа мышечных ядер и базофилией мышечных элементов. В последующем развивается интерстициальный фиброз.
Классификация
В настоящее время используется классификация (по R.L. Woltman, 1994), в которую вошли идиопатические воспалительные миопатии и миопатии, вызываемые инфекциями, лекарствами и токсинами.
- Идиопатические воспалительные миопатии:
- Первичный полимиозит.
- Первичный дерматомиозит.
- Ювенильный дерматомиозит.
- Миозит, ассоциирующийся с опухолями.
- Миозит с "включениями".
- Миозит, ассоциирующийся с эозинофилией.
- Локализованный или очаговый миозит.
- Оссифицирующий миозит.
- Гигантоклеточный миозит.
- Миопатии, вызываемые инфекциями.
- Миопатии, вызываемые лекарственными средствами.
Симптомы
Первичный полимиозит. Может начинаться внезапно или постепенно. Вначале развивается симметричная мышечная слабость проксимальных мышц конечностей, особенно бедер. Больным трудно подниматься и спускаться по лестнице, вставать из положения на корточках. При поражении мышц плечевого пояса больные испытывают затруднение при подъеме рук вверх и причесывании. Нередко поражаются мышцы шеи, что сопровождается нарушением ее сгибания. У многих больных развивается слабость поперечнополосатых мышц глотки и верхней трети пищевода, приводящая к дисфагии. У некоторых больных поражаются дыхательные мышцы, что приводит к тяжелым дыхательным нарушениям. Дистальные мышцы конечностей поражаются сравнительно редко. Атрофия мышц и контрактуры могут развиться на поздних стадиях заболевания. Примерно у 35% больных имеются поражения сердечной мышцы, проявляющиеся инфарктоподобными изменениями на ЭКГ, аритмиями и сердечно-сосудистой недостаточностью. У некоторых больных развиваются артралгии и синдром Рейно.
Первичный дерматомиозит. Составляет треть случаев дерматомиозита-полимиозита. Поражение мышц развивается у большинства больных одновременно с изменениями кожи. На коже появляются локальные или диффузные эритематозные участки с характерным лиловым оттенком, шелушением и атрофией. Высыпания чаще всего локализуются на веках, переносице, щеках, лбу, над локтевыми, коленными и пястнофаланговыми суставами и распространяются на шею и верхнюю часть туловища. Нередко выявляется синдром Готтрона (шелушение и атрофия кожи, наиболее выраженная на кистях и стопах), гелиотропные ("солнечные") веки (припухшие и имеющие фиолетовое окрашивание) и "руки мастерового" (эритематозные гипертрофические изменения кожи ладоней и пальцев).
Поражения суставов. Иногда у больных развиваются симметричные воспалительные поражения мелких суставов кистей и лучезапястных суставов. Но деструктивные изменения суставов обычно отсутствуют. Воспаление носит преходящий характер и быстро купируется при лечении глюкокортикоидами.
Поражения легких. Чаще всего проявляются интерстициальными пневмониями, альвеолитом и медленнопрогрессирующим фиброзом, обусловленными антителами к синтетазам транспортных РНК. Проявляются эти поражения непродуктивным кашлем и прогрессирующей дыхательной недостаточностью. Поражение диафрагмальных мышц и развивающаяся сердечная недостаточность усугубляют смешанную одышку.
Поражение почек. Развивается редко и проявляется протеинурией и нефротичексим синдромом. Иногда вследствие выраженной миоглобинурии развивается острая почечная недостаточность.
Дерматомиозит и полимиозит при злокачественных новообразованиях. Примерно у 10–25% больных выявляют онкологические заболевания, главным образом, у взрослых старше 40 лет. Особенно высок риск у онкологических больных старше 60 лет. Миопатии чаще всего развиваются при раке легкого, ЖКТ, молочной железы, матки, яичников и лимфопролиферативных заболеваниях. Данные формы дерматомиозита–полимиозита рассматривают как самостоятельные и относят к паранеопластическим синдромам. Для исключения злокачественных новообразований проводят тщательное клиническое, гинекологическое, ректальное и по показаниям рентгенологическое исследование, общий и биохимический анализ крови, цитологическое исследование мокроты и мочи. Дополнительно рекомендуют определение простатспецифического антигена у мужчин и СА-125 у женщин (антиген опухоли яичника). Прогноз неблагоприятный, больные погибают от основного заболевания, осложнений или не поддающегося лечению поражения мышц.
Ювенильный дерматомиозит. Среди больных дерматомиозитом примерно 25% составляют дети, у которых наблюдается клиническая и гистологическая картина васкулита с поражением кожи, мышц, ЖКТ, почек и других органов. Нередко развиваются очаги некроза кожи, ишемические инфаркты мышц, почек и ЖКТ. При прогрессировании заболевания развиваются контрактуры вследствие кальцификации мышц и околосуставной подкожной клетчатки. Прогноз заболевания такой же, как у взрослых. Но существует мнение, что у детей прогноз лучше, чем у взрослых. Возможно, эта противоречивость обусловлена различием диагностических критериев ювенильного дерматомиозита.
Миозит с "включениями". Данная форма относится к воспалительным миопатиям, носит спорадический характер и по клинике напоминает полимиозит. Болезнь развивается обычно у пожилых мужчин, проявляется очаговым порождением как проксимальных, так и дистальных мышц конечностей и имеет медленное прогрессирование. Характерно для миозита с "включениями" раннее появление выраженной слабости четырехглавой мышцы, сгибателей пальцев и запястья. Типичный гистологический признак, выявляемый в биоптате мышц, – вакуоли, окруженные ободком из базофильных гранул, содержащих амилоид. С помощью электронной микроскопии обнаруживают в саркоплазме и ядрах включения, напоминающие по форме нуклеокапсиды парамиксовирусов. В сыворотке крови отсутствуют аутоантитела. Характерна резистентность к глюкокортикоидам и другим иммуносупрессивным препаратам. Через 5–10 лет при хроническом прогрессировании течения больные утрачивают способность к самостоятельному передвижению.
Дерматомиозит и полимиозит, ассоциированный с ревматическими болезнями. Поражение проксимальных мышц, идентичное полимиозиту, может быть при системной склеродермии, ревматоидном артрите, системной красной волчанке и реже при узелковом периартериите и ревматизме. Но чаще всего миозит является компонентом смешанного заболевания соединительной ткани. На долю этой формы дерматомиозита–полимиозита приходится 20% всех перекрестных синдромов, для которых характерны полиартрит и феномен Рейно, высокий титр антинуклеарных аутоантител и повышение активности КФК. Биопсия мышц не всегда позволяет привести дифференциальную диагностику, так как выявляемые патологические изменения в мышцах сходны при полимиозите и других заболеваниях соединительной ткани. Как правило, хорошо поддается лечению глюкокортикоидами.
Диагностика
Для всех форм полимиозита характерно повышение активности мышечных ферментов (КФК, альдолазы, ЛДГ, АСЛ и АЛТ), выраженность которой коррелирует со степенью активности заболевания. Показатели общего анализа крови нормальные. У большинства (80–90%) больных выявляют аутоантитела к ядерным и цитоплазматическим антигенам. Наличие аутоантител к НEр-2 клеткам является стандартным диагностическим тестом.
Все воспалительные миопатии сопровождаются образованиями специфических аутоантител. К ним относятся аутоантитела к синтетазам транспортной РНК (антитела J0-1 к гистидил-тРНК-синтетазе) и распознающим сигналы частицам, взаимодействующим с нуклеопротеинами миозина. При сочетании полимиозита с системной склеродермией обнаруживают антитела к Sel-70, a с СКВ и синдромом Шёгрена – к Sm, Ro/SS-A La/SS-В.
При регистрации электромиограмм у большинства больных (80%) выявляют типичную миопатическую триаду: мелкие, короткие и полифазные (более чем из четырех фаз) потенциалы действия двигательных единиц. При гистологическом исследовании биоптата мышц выявляют очаговый некроз, дегенерацию и фагоцитоз мышечных волокон и воспалительные инфильтраты, состоящие из лимфоцитов, макрофагов и плазматических клеток. В скелетных мышцах развивается регенерация с базофилией мышечных элементов, увеличением числа и величины мышечных ядер. По периферии мышечных пучков развивается атрофия мышечных волокон, обусловленная некрозом эндотелия и уменьшением количества капилляров. При хроническом течении заболевания формируются интерстициальные фиброзные инфильтраты.
Диагностические критерии были разработаны A. Bohan et J.B. Petter в 1975 г. Эти критерии следует дополнить пунктами о наличии недеструктивного артрита и обнаружении аутоантител J0-1. При обнаружении одного типа поражения кожи и четырех других признаков из пунктов 1–4 и аутоантител J0-1 диагноз будет достоверен (чувствительность – 98,9%, специфичность – 95,2%).
Диагностические критерии дерматомиозита–полимиозита (по А. Bohan, J.B. Petter, 1975):
- Симметрическая слабость мышц плечевого и тазового пояса и передних сгибателей шеи, прогрессирующая в течение нескольких недель или месяцев в сочетании или при отсутствии дисфагии или поражения дыхательной мускулатуры.
- При гистологическом исследовании мышц признаки некроза мышечных фибрилл 1-го и 2-го типов, фагоцитоз, регенерация с базофилией, крупные ядра и ядрышки в сарколемме, перифасциальная атрофия, вариабельность размера миофибрилл, воспалительный экссудат.
- Повышение сывороточной концентрации мышечных ферментов (КФК, альдолаза, АСТ, АЛТ и ЛДГ).
- Электромиографические изменения: короткие, мелкие полифазные моторные единицы, фибрилляции и т.д.
- Дерматологические проявления, включающие гелиотропную окраску кожи век с периорбитальным отеком; чешуйчатый эритематозный дерматит на тыльной поверхности кистей, особенно над пястно-фаланговыми и проксимальными межфаланговыми суставами (симптом Готтрона) и поражение кожи над коленными, локтевыми суставами, лица, шеи, верхней половины грудной клетки.
Генерализованная мышечная слабость или слабость проксимальных мышц в сочетании с типичными кожными изменениями являются характерными признаками дерматомиозита–полимиозита. Но мышечная слабость, сопровождающаяся или не сопровождающаяся миалгиями, может быть при многих других заболеваниях.
Лечение
Основными препаратами при лечении тяжелого дерматомиозита–полимиозита являются глюкокортикоиды (преднизолон, метилпреднизолон). Начальная доза глюкокортикоидов колеблется от 1 до 2 мг/кг/сут, которую в течение 2–3 нед принимают 3 раза в день, затем – один раз в утренние часы. Улучшение наступает в течение 3–4 нед лечения, но у некоторых больных оно наступает через 3 мес. Отсутствие эффекта к этому времени являются основанием для увеличения дозы глюкокортикоидов.
После улучшения состояния больных, нормализации мышечной силы и КФК суточную дозу преднизолона уменьшают на 5 мг каждые 4 нед с обязательным регулярным контролем мышечной силы и активности КФК в сыворотке. Но следует учитывать то, что преднизолон в начале лечения может снижать активность КФК без уменьшения воспаления и инфильтрации мышц. Для уменьшения побочных эффектов больших доз глюкокортикоидов необходимо сочетать их с приемом Н 2 -блокаторов, витамина Д и препаратов кальция. После снижения суточной дозы до 40 мг переходят на прием 80 мг препарата через день. Если развивается рецидив заболевания дозу препарата вновь увеличивают до стойкого улучшения.
При неэффективности лечения в течение 3 мес высокими дозами глюкокортикоидов рекомендуют назначение азатиоприна в дозе 2–3,5 мг/кг/сут в 2–3 приема. Эффект развивается через 4–5 мес, показателем эффективности является снижение числа лимфоцитов до 750 мкл -1 . В последние годы используют метотрексат в начальной дозе 10 мг/нед с последующим ее постепенным повышением до 0,5 мг/кг/нед. Сочетанная терапия этих препаратов с глюкокортикоидами позволяет снизить дозу последних. Циклофосфамид и циклоспорин менее эффективны и их применяют при развитии интерстициального фиброза легких. Но все эти препараты угнетают кроветворение, вызывают нарушения ЖКТ и повышают риск инфекций.
По данным последних исследований, при неэффективности глюкокортикоидов улучшение может быть достигнуто с помощью в/в введения нормального иммуноглобулина в дозе 0,4 г/кг/сут в течение 5 сут. Применение плазмафереза показано больным с тяжелыми формами дерматомиозита–полимиозита, которым проводится лечение сочетанием глюкокортикоидов с азатиоприном или метотрексатом.
Очень важное значение имеет реабилитация больных с помощью физиотерапии, ЛФК и вспомогательных приспособлений. В острую фазу заболевания больной с помощью методиста выполняет пассивные упражнения и напряжение мышц, при улучшении состояния показаны изометрические и изотонические упражнения для восстановления мышечной силы.
Прогноз
Лечение большинства больных эффективно, но общая смертность при дерматомиозите-полимиозите в 4 раза выше, чем среди населения в целом. Пятилетняя выживаемость составляет 80%, у детей она еще выше.
Прогноз хуже у женщин и пожилых больных, при остром и тяжелом течении заболевания (высокая лихорадка, выраженная дисфагия, поражение внутренних органов), поздней диагностике и неадекватном лечении, наличии в сыворотке антител к J0-1 и злокачественных новообразованиях.
Наличие полимиозита даже при отсутствии значительного повышения лабораторных признаков активности требует назначения относительно высоких доз кортикостероидов (40—50 мг преднизолрна). Большинство из этих больных теперь рассматриваются как лица со смешанным заболеванием соединительной ткани.
Поражение костного аппарата связано не столько с суставной патологией, сколько с сосудистотрофическими нарушениями.
Это явления остеолиза или резорбции кости [Khafistanteen I. et al., 1988], описанные впервые Г. И. Мещерский (1909) как мутилирующая склеродактилия. Наиболее характерен остеолиз ногтевых фаланг (рис. 5.4, А, Б), который отмечен нами у 40% больных. Мы наблюдали у отдельных больных также рассасывание средних и основных фаланг с образованием секвестров или полным исчезновением костной ткани, части шиловидного отростка, верхней челюсти и даже ребер.

Рис. 5.4. Остеолиз ногтевых фаланг пальцев рук (А) и ног (Б).
В общей патологии рассасывание ногтевых фаланг встречается лишь в отдельных случаях таких относительно редких заболеваний, как синдром Элерса — Данло, семейный остеолиз, сирингомиелия, подагра, лепра, которые легко дифференцируются от склеродермии по общей картине заболевания.
Генез остеолиза при ССД полностью не выяснен, хотя большинство ученых связывают его с ухудшением локального кровоснабжения. При этом следует учитывать как нарушения питания в связи с поражением сосудов, так и общетрофические сдвиги, свойственные заболеванию, а в качестве одного из возможных механизмов нарушения структуры кости — изменения в собственно коллагеновой матрице костной ткани.
При ССД нередко наблюдается подкожный и внутрикожный кальциноз с преимущественной локализацией в области ногтевых фаланг кисти, метакарпофаланговых суставов, предплечий, локтевых и коленных суставов, нередко по ходу фасций и сухожилий (рис. 5.5). У отдельных больных выявлен частичный кальциноз надгортанника, голосовых связок, перикарда, мышцы и клапанного аппарата сердца, капсулы печени и селезенки. Величина кальциевых отложений варьирует от точечных до больших конгломератов.

Рис. 5.5. Кальциноз в области кистей у больной системной склеродермией.
Известный первоначально как синдром Тибьержа — Вейссенбаха, по имени авторов, впервые описавших кальциноз при склеродермии в 1910 г., в настоящее время он рассматривается как один из основных компонентов CREST-синдрома (С-кальциноз); более характерен для хронического течения болезни.
Кальциноз при клиническом и рентгенологическом исследовании выявлен нами у 23% больных: у 1/3 из них было подострое, а у 2/3 — хроническое течение болезни, длительность которого колебалась от 5 до 29 лет. У большинства больных отмечены развернутая картина ССД или уже далеко зашедшие проявления болезни. Наблюдения показали, что при длительности заболевания до 5 лет кальциноз развивается очень редко, а затем частота его нарастает по мере увеличения продолжительности болезни и после 10 лет составляет 50% и более.
Кальцинация тканей чаще происходит постепенно и выявляется лишь рентгенологически, а при локализации кальциноза в пальцах — иногда по деформации последних. При более бурном (чаще по типу отдельных обострении) развитии процесса отмечается воспалительная инфильтрация тканей с выраженным болевым синдромом, ухудшением общего состояния и иногда лихорадочной реакцией.
При поверхностном расположении очаги кальциноза могут вскрываться, при этом выделяется белая крошковатая или жидкая масса. Подобное отторжение наряду с возможностью резорбции небольших депозитов кальция полибластами и эффективным лечением, препятствующим альтерации тканей, обусловливают положительную динамику процесса и исчезновение отдельных очагов кальциноза.
При резко выраженном кальцинозе с преимущественной локализацией его в подкожной клетчатке и (или) мышцах необходимо исключить дерматомиозит, полимиозит (возможно, как перекрестный синдром), оссифицирующий миозит и идиопатический кальциноз.
Таким образом, суставно-костно-мышечный синдром при ССД имеет своеобразную клинико-рентгенологически-морфологическую картину, что при учете большой частоты позволяет считать его одним из основных диагностических признаков болезни. Поражение легких — одна из характерных висцеральных локализаций склеродермического процесса; колеблется от 30 до 90%, причем наибольший процент поражения выявляют при функциональном и морфологическом методах исследования.
В основе легочной патологии при ССД лежит интерстициальное поражение легких с развитием фиброза, утолщением альвеолярных стенок и нарушением диффузии газов через измененную мембрану, утолщение интимы прекапиллярных артериол и других сосудов с сужением и даже полной облитерацией просвета их. Характерно преимущественное развитие пневмофиброза в базальных отделах легких, нередко поражение плевры. Преобладание сосудистой патологии ведет к развитию легочной гипертензии.
С помощью сканирования легких и бронхоальвеолярного лаважа показано, что развитию пневмофиброза предшествуют воспалительные изменения с картиной альвеолита. При бронхоальвеолярном лаваже обнаружено повышенное содержание клеток (нейтрофилы, эозинофилы, иногда —лимфоциты), потенциальных медиаторов воспаления (коллагеназа) и иммунных комплексов, коррелировавших с количеством нейтрофилов в лаважной жидкости.
Значительное превышение у отдельных больных уровня (в 45 раз) и частоты обнаружения иммунных комплексов в бронхоальвеолярной жидкости по сравнению с сывороткой крови позволило предположить их локальное образование и избирательное отложение в нижних отделах дыхательного тракта при ССД.
Клиническая картина варьирует в зависимости от выраженности процесса. Наиболее частые симптомы поражения легких при ССД — одышка, затруднение глубокого вдоха и снижение экскурсии нижнего легочного края. Одышка может быть обусловлена увеличением числа дыханий вследствие снижения легочной эластичности и гипервентиляцией в связи с фиброзными изменениями в бронхиальных стенках и сосудах легких, расширением воздушных путей и неадекватностью вентиляционно-перфузионных соотношений.
Легочная гипертензия часто сопровождается гипервентиляцией, особенно при нагрузке. Естественно, при склеродермическом поражении сердца и почек также возникает одышка. Роль индуративных изменений кожи и ригидности грудной клетки в происхождении одышки, по-видимому, невелика.
Боли в грудной клетке относительно редки и, как правило, связаны с плевритом, кардиальной патологией или невралгиями. Кашель, чаще сухой, обусловлен развитием бронхита и бронхоэктазов.
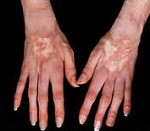
Системная склеродермия - диффузная патология соединительной ткани, для которой характерны фиброзно-склеротические изменения кожи, суставно-мышечного аппарата, внутренних органов и сосудов. Типичными признаками системной склеродермии служат синдром Рейно, уплотнение кожи, маскообразное лицо, телеангиэктазии, полимиозит, суставные контрактуры, изменения со стороны пищевода, сердца, легких, почек. Диагностика системной склеродермии основана на комплексе клинических данных, определении склеродермических аутоантител, биопсии кожи. Лечение включает антифиброзные, противовоспалительные, иммуносупрессивные, сосудистые средства, симптоматическую терапию.
- Причины
- Классификация
- Симптомы системной склеродермии
- Диагностика
- Лечение системной склеродермии
- Прогноз
- Цены на лечение
Общие сведения
Причины
Точные представления о причинах системной склеродермии отсутствуют. Накопленные наблюдения позволяют лишь высказывать отдельные этиологические гипотезы. В пользу генетической детерминированности свидетельствуют факты семейной истории системной склеродермии, а также наличие у ближайших родственников других склеродермических болезней, коллагенозов (СКВ, ревматоидного артрита, синдрома Шегрена), микроангиопатий, кардиопатий и нефропатий неизвестного генеза. Выявлена ассоциация склеродермии с определенными антигенами и аллелями HLA-системы, определяющими иммунный ответ, что также свидетельствует о наличии генетического следа в генезе патологии.
Наряду с наследственной теорией, широко обсуждается роль инфекции, в первую очередь цитомегаловирусной. Некоторые пациенты связывают дебют заболевания с перенесенным гриппом или стрептококковой ангиной. Ряд наблюдений указывает на триггерную роль химических агентов: кварцевой и угольной пыли, растворителей, лекарственных средств (в частности, блеомицина и других цитостатиков). Доказано участие вибрационного воздействия, стресса, охлаждения и обморожения, травм в запуске иммунопатологических сдвигов при системной склеродермии. Фоном для развития системного склероза может служить гормональная перестройка, обусловленная пубертатом, родами, абортом, климаксом. У отдельных пациентов началу заболевания предшествуют операции (удаление зуба, тонзиллэктомия и др.) и вакцинация. Т. о., на основании имеющихся данных можно сделать вывод о мультифакториальном генезе системной склеродермии, сочетающем в себе сложное взаимодействие эндо- и экзогенных факторов с наследственной предрасположенностью.
Патогенетические механизмы системной склеродермии изучены лучше этиологии. Ключевую роль в них играют нарушения клеточного и гуморального иммунитета, приводящие к повышению числа CD4 + и В-лимфоцитов, и реакция гиперчувствительности, обусловливающая образование широкого спектра аутоантител (антинуклеарных, антицентромерных, анти-Scl-70, антинейтрофильных, антиэндотелиальных, цитоплазматических, АТ к соединительной ткани и др.) и циркулирующих иммунных комплексов. Подобная иммунная активация способствует гиперактивности фибробластов и повреждению сосудистого эндотелия. Специфика заболевания определяется генерализованным склерозом органов и тканей (кожи, костно-суставной и мышечной системы, ЖКТ, сердца, легких, почек) и развитием облитерирующей микроангиопатии. Рассмотренный механизм позволяет отнести системную склеродермию к аутоиммунным заболеваниям.
Классификация
Системная склеродермия (диффузная или генерализованная склеродермия, прогрессирующий системный склероз) может протекать в нескольких клинических формах:
- Пресклеродермия не имеет дерматологических проявлений и сопровождается только феноменом Рейно.
- Для диффузной склеродермии патогномонично стремительное развитие, поражение кожи, сосудов, мышечно-суставного аппарата и внутренних органов в течение первого года заболевания.
- Лимитированная форма протекает с медленно развивающимися фиброзными изменениями, преимущественным поражением кожных покровов и поздним вовлечением внутренних органов.
- При склеродермии без склеродермы отмечаются только висцеральные и сосудистые синдромы без типичных кожных проявлений.
- Перекрестная форма может проявляться сочетанием системной склеродермии с дерматомиозитом, полимиозитом, СКВ, РА, васкулитами.
Системная склеродермия может протекать в хронической, подострой и острой форме. При хроническом течении на протяжении многих лет единственным указанием на заболевание служит синдром Рейно; другие типичные поражения развиваются постепенно и длительно. При подостром варианте системной склеродермии преобладает кожно-суставной (склеродермия, полиартрит, полимиозит) и висцеральный синдром (сердечно-легочный) при незначительных вазомоторных нарушениях. Острая форма патологии отличается стремительным (в течение 12 мес.) формированием системного фиброза и микрососудистых нарушений. Различают три степени активности системной склеродермии: I – минимальную, типичную для хронического варианта; II – умеренную, обычно встречающуюся при подостром процессе; III – максимальную, сопровождающую течение острой и иногда подострой форм.
Симптомы системной склеродермии
Клиническая специфика системной склеродермии заключается в полиморфности и полисиндромности проявлений. Варианты развития болезни могут варьировать от маловыраженных форм с относительно благоприятным прогнозом до быстропрогрессирующих диффузных поражений с ранним фатальным исходом. В дебюте системной склеродермии, еще до появления специфических поражений, отмечается потеря веса, слабость, субфебрилитет.
Наиболее ранним признаком заболевания служит синдром Рейно, характерный для 99% пациентов и протекающий с преходящими пароксизмами вазоспазма. Под воздействием стресса или охлаждения пальцы рук резко бледнеют, затем кожа приобретает синевато-фиолетовую окраску. Сосудистый спазм может сопровождаться чувством зябкости и онемения кистей. После разрешения вазоконстрикции наступает стадия реактивной гиперемии: кожа становится ярко-розовой, появляется ощущение ломоты и боли в пальцах. Феномена Рейно при склеродермии может носить системный характер, т. е. распространяться на сосуды кожи лица, языка, почек, сердца и др. органов.
Кожный синдром присутствует у большинства больных системной склеродермией. В своей эволюции он проходит 3 фазы: воспалительного отека, уплотнения (индурации) и атрофии кожи. Начальную стадию характеризует появление плотного отека кожи рук и ног, сопровождающегося зудом. В дальнейшем развивается склеродактилия (утолщение кожи пальцев), образуются трофические язвы, деформируются ногти. Лобные и носогубные складки сглаживаются, в результате чего лицо приобретает маскообразное выражение. Вследствие атрофии сальных и потовых желез кожа становится сухой и грубой, лишенной волосяного покрова. Часто обнаруживаются телеангиэктазии, депигментация или гиперпигментация кожи, подкожные кальцинаты.
Мышечно-суставной синдром также часто сопутствует системной склеродермии. Типичны отечность и скованность суставов, артралгии – данный симптомокомплекс носит название склеродермического полиартрита. Вследствие уплотнения кожи формируются сгибательные контрактуры суставов, развиваются теносиновиты. Возможен остеолиз ногтевых фаланг, приводящий к укорочению пальцев. Поражение мышц при системной склеродермии протекает по типу полимиозита или невоспалительной миопатии.
Висцеральные поражения могут затрагивать ЖКТ (90% случаев), легкие (70%), сердце (10%), почки (5%). Со стороны органов пищеварения отмечается дисфагия, изжога, тошнота и рвота. Развивается рефлюкс-эзофагит, усугубляющийся образованием язв и стриктур пищевода. На этом фоне у больных системной склеродермией повышен риск формирования пищевода Барретта и аденокарциномы. При поражении тонкого кишечника возникает диарея, метеоризм, похудание; при вовлечении толстого кишечника – запоры и кишечная непроходимость.
Поражение легких при системной склеродермии может выражаться в виде пневмофиброза и легочной гипертензии. Оба синдрома проявляются непродуктивным кашлем, прогрессирующей экспираторной одышкой и дыхательной недостаточностью. Поражение легких служит ведущей причиной летальных исходов у больных системной склеродермией, поэтому расценивается как прогностически неблагоприятный фактор. При вовлечении сердца могут развиваться аритмии, перикардит (адгезивный или экссудативный), эндокардит, сердечная недостаточность.
Почечный синдром при системной склеродермии чаще протекает в форме латентной нефропатии с умеренными функциональными нарушениями. Однако у ряда больных в первое пятилетие от дебюта заболевания развивается грозное, потенциально летальное осложнение - острая склеродермическая почка, которая протекает с гиперренинемией, злокачественной артериальной гипертензией, тромбоцитопенией и гемолитической анемией, стремительно нарастающей почечной недостаточность. В числе прочих синдромальных проявлений системной склеродермии встречаются полиневропатия, синдром Шегрена, аутоиммунный тиреоидит, первичный билиарный цирроз печени и др.
Диагностика
Американской коллегией ревматологов разработаны критерии, на основании которых может быть выставлен диагноз системной склеродермии. Среди них выделяют большой критерий (проксимальная склеродермия - уплотнение кожи кистей рук, лица и туловища) и малые (склеродактилия, дигитальные рубчики, двусторонний пневмофиброз). При выявлении двух малых или одного большого признака клинический диагноз можно считать подтвержденным. Дифференциально-диагностические мероприятия проводятся как внутри группы склеродермических болезней, так и среди других системных заболеваний: синдрома Шегрена, полимиозита, дерматомиозита, облитерирующего тромбангиита и мн. др.
Общеклинические анализы малоинформативны, а выявляемые в них изменения – неспецифичны. Со стороны крови отмечается гипохромная анемия, лейкоцитопения или лейкоцитоз, умеренное увеличение СОЭ. В общем анализе мочи может выявляться протеинурия, лейкоцитурия, микрогематурия. Биохимические показатели указывают на признаки воспаления (повышение уровня серомукоида и фибриногена, СРБ, РФ). Наибольшее значение имеют результаты иммунологического обследования. При системной склеродермии в крови обнаруживаются склеродермические аутоантитела-маркеры: АТ к Scl-70 и антицентромерные АТ.
Среди инструментальных методик для ранней диагностики системной склеродермии наибольшую ценность представляет капилляроскопия ногтевого ложа, позволяющая выявить начальные признаки болезни. Для оценки состояния костной системы проводится рентгенография кистей. С целью выявления интерстициального пневмофиброза целесообразно выполнение рентгенографии и КТ легких. Для исследования ЖКТ назначают рентгенографию пищевода, рентгенографию пассажа бария по кишечнику. Электрокардиография и ЭхоКГ необходимы для выявления кардиогенных поражений и легочной гипертензии. Электромиография позволяет подтвердить миопатические изменения. Для гистологической верификации системной склеродермии проводится биопсия кожи, мышц, почек, легких, перикарда.
Лечение системной склеродермии
Лицам, страдающим системной склеродермией, следует избегать стрессовых факторов, вибрации, переохлаждения, инсоляции, контакта с бытовыми и производственными химическими агентами, отказаться от курения и употребления кофеина, приема сосудосуживающих средств. Фармакотерапия, ее дозировки и длительность зависят от клинической формы, активности и скорости прогрессирования заболевания, тяжести висцеральных поражений.
Патогенетическая терапия системной склеродермии проводится с использованием сосудистых, антифиброзных и иммуносупрессивных препаратов. Для предупреждения эпизодов сосудистого спазма и профилактики ишемических осложнений назначаются вазодилататоры (нифедипин, верапамил, дилтиазем, циннаризин и др.), антиагреганты (ацетилсалициловую кислоту, пентоксифиллин) и антикоагулянты (гепарин, варфарин). С целью подавления развития системного фиброза используется D-пеницилламин. Противовоспалительная терапия при системной склеродермии включает прием НПВП (ибупрофен, диклофенак, нимесулид) и глюкокортикоидов. Препараты данной группы помогают уменьшить признаки воспаления (миозита, артрита, тендосиновита) и иммунологическую активность. Для замедления прогрессирования системного фиброза может применяться метотрексат, циклоспорин, пульс-терапия циклофосфаном.
Симптоматическая терапия при системной склеродермии направлена на уменьшение расстройств пищеварения, сердечной недостаточности, легочной гипертензии. При развитии склеродермического почечного криза назначается каптоприл, эналаприл; в некоторых случаях может потребоваться проведение гемодиализа. Хирургическое лечение – грудная симпатэктомия - показано при осложненной форме синдрома Рейно.
Прогноз
Прогноз при системной склеродермии в целом неблагоприятный. Самая низкая пятилетняя выживаемость (30-70%) ассоциируется с диффузной формой. Предикторами неблагополучного прогноза выступают легочный и почечный синдромы, дебют болезни у пациентов старше 45 лет. Лимитированная форма и хроническое течение болезни имеют более благоприятный прогноз и лучшую выживаемость, при них возможно планирование беременности и благополучное родоразрешение. Пациенты с системной склеродермией подлежат диспансерному учету и наблюдению каждые 3–6 месяцев.
Читайте также: