
Прогрессирующая перонеальная мышечная атрофия
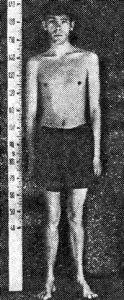
Перонеальная мышечная атрофия, болезнь Шарко—Мари—Рота — медленно прогрессирующее наследственное заболевание, проявляющееся в основном атрофия ми мышц дистальных отделов конечностей.
Приблизительно в половине наблюдений она носит наследственно-семейный характер: известны случаи заболевания в 4 и даже в 5—6 поколениях одной семьи или только у братьев и сестер, т. е. болезнь может передаваться и по аутосомно-доминантному (с высокой пенетрантностью, около 80%) и реже —по аутосомно-рецессивкому типам. По клиническому течению спорадические случаи (вероятно, в их основе чаще всего лежит рецессивный тип передачи) ничем не отличаются от семейных. Заболевание начинается в детском или юношеском возрасте.
Патоморфологически наблюдаются перерождение осевых цилиндров и миелиновой оболочки периферических нервов и нервных корешков, атрофии клеток передних рогов, главным образом в поясничной и шейной областях спинного мозга. Изменения в мышцах (атрофия, дегенерация и распад мышечных волокон, гиперплазия интерсти-циальной соединительной ткани и др.) носят ггреимущест-венно вторичный неврогенный характер. Наряду с этим возможно вовлечение и проводящих систем спинного мозга (боковых и задних канатиков).
Патогенез обусловлен нейротрофическими расстройствами в мышцах вследствие поражения передних корешков и периферических нервов, а также передних рогов.
Клиническая картина. Основной симптом заболевания—симметричные атрофии дистальных отделов конечностей, которые начинаются обычно с ног. Поражаются межкостные мышцы, разгибатели пальцев и стоп, затем перонеальная группа мышц, а также мышцы бедра. Стопа несколько свисает, пальцы приобретают когтевидную установку (вогнутая стопа — рех excavatus).
Нередко эти изменения сочетаются с костной аномалии.
по типу так называемой стопы Фридрейха — она несколько укорочена и имеет высокий свод (см. рис. 64).
В данном случае, хотя клиника, данные ЭМГ и наследственность (наличие больных в трех поколениях семьи) типичны для невральной амиотрофии, диагноз был установлен только через 2 года после появления первых симптомов заболевания. Обращает на себя внимание благоприятное течение невральной амиотрофии (отец и дед пробанда дожили до преклонного возраста).
Диагноз и дифференциальный диагноз. Распознавание болезни основано на данных анамнеза (начало в детском или юношеском возрасте, случаи заболевания в семье), характерных симптомах (атрофии голеней и предплечий, стопа Фридрейха и др.), данных электромиографии (выявляются признаки невритического, переднерогового и супра-сегментарного типов нарушения мышечного электрогенеза) и исследования скорости проведения возбуждения по нервным стволам (она резко снижается). Не враль ну ю амио-трофию необходимо разграничивать с ее подвидом —гипертрофическим ин-терстициальным невритом Дежерина— С о т т а, который отличают значительно выраженные утолщения периферических нервных стволов, атаксия, нистагм, зрачковые нарушения (миоз, анизокория), а также с дис-тальной миопатией и полиневропатией.
Лечение симптоматическое и общеукрепляющее. Применяют витамины группы В (В1 и В6), АТФ, церебролизин, стрихнин, пантокрин, стекловидное тело, антихолинэстераз-ные препараты (галантамин, оксазил), физиотерапию (ванны, грязелечение), массажу Терапию проводят повторными 1— 2-месячными курсами после 2—3-месячного перерыва.
Обязательны систематические занятия лечебной гимнастикой. При наличии свисающих стоп и затруднении ходьбы показана ортопедическая помощь (специальная обувь, тенотомия).
Прогноз. Течение заболевания медленно прогрессирующее. Процесс длительно (10—15 лет) ограничивается вовлечением нижних конечностей, затем постепенно развиваются атрофии и верхних конечностей — кисти и предплечья. Мышцы туловища, шеи, головы и проксимальных отделов конечностей обычно не страдают. Иногда небольшие атрофии могут быть в нижней части бедер и области плеч. У ряда больных, особенно при условии систематического лечения, состояние в течение многих лет может оставаться стабильным.
Статья на тему Невральная амиотрофия
Перонеальная мышечная атрофия (болезнь Шарко-Мари-Тута) - это заболевание представляет собой наиболее распространенную форму генетически детерминированных невропатий; распространенность 3,8:100 000 населения. Тип наследования аутосомно-доминантный, экспрессивность составляет 83 %. Аномальный ген картирован в локусе 17р11.2. Продукт гена — протеин Р22 периферического миелина (РМР22). Встречающаяся значительно реже НМСН типа I с аутосомно-рецессивным типом наследования возникает в результате дефекта гена в локусе Xql3.1, что приводит к нарушению синтеза белка коннексин-32.
Клинические проявления болезни Шарко-Мари-Тута у детей. У большинства пациентов симптомы заболевания отсутствуют до позднего детского или раннего подросткового возраста. Однако у некоторых детей походка нарушается уже на втором году жизни. Большеберцовый и малоберцовый нервы поражаются наиболее рано, и их поражение наиболее выражено по сравнению с другими нервами. Дети с этим заболеванием кажутся неуклюжими, они часто спотыкаются и падают. Дебют симптомов может быть отсрочен до 6-го десятилетия жизни.
Мышцы передней поверхности голени атрофированы, и нога принимает характерную форму ноги аиста. Мышечная атрофия сопровождается прогрессирующей слабостью этих мышц, в связи с чем тыльное сгибание стопы затруднено, впоследствии формируется симптом свисающей стопы. Поражение билатеральное, но может определяться легкая асимметрия. Возможна деформация pes cavus (когтистая лапа) в связи с денервацией внутренних мышц стопы, что приводит к еще большему нарушению походки. Атрофия мышц предплечий и кистей обычно выражена не настолько сильно, как атрофия мышц нижних конечностей, при прогрессировании заболевания возможна контрактура лучезапястных суставов и пальцев рук, что приводит к формированию когтистой лапы. Слабость проксимальных мышц возникает на поздних стадиях заболевания и обычно умеренно выражена. Аксиальные мышцы йе поражаются.
Перонеальная мышечная атрофия медленно прогрессирует на протяжении всей жизни, однако в некоторых случаях двигательные нарушения быстро прогрессируют в течение нескольких лет. У большинства пациентов сохраняется способность к ходьбе и нормальная продолжительность жизни, хотя необходимо применение ортопедических приспособлений для стабилизации голеностопных суставов.
Среди чувствительных волокон поражаются главным образом крупные миелинизированные нервные волокна, передающие информацию о проприоцептивной и вибрационной чувствительности, возможно и повышение порога болевой и температурной чувствительности. Некоторые дети жалуются на ощущение покалывания или жжения в стопах, боль возникает редко. В связи с уменьшением мышечной массы нервы особенно уязвимы к травме или компрессии. Вегетативные проявления могут включать нарушение вазомоторного контроля с появлением красных пятен или бледности кожи стоп, при этом стопы необычно холодные на ощупь.
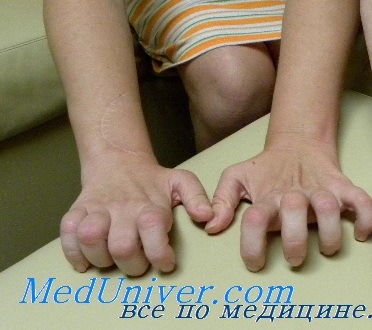
При пальпации нервы часто утолщены. Сухожильные рефлексы в дистальных отделах конечностей утрачены. Черепные нервы не поражаются. Функция тазовых органов не нарушена. При вегетативной невропатии отсутствует поражение сердца, ЖКТ и мочевого пузыря. Интеллект не снижен.
Синдром Давиденкова — вариант НМСН типа I с лопаточно-перонеальной атрофией и слабостью мышц.
Скорость проведения возбуждения по двигательным и чувствительным нервам значительно снижена, иногда достигает всего лишь 20 % нормального значения. При выявлении новых случаев заболевания при отсутствии семейного анамнеза необходимо обследование обоих родителей и исследование скорости проведения возбуждения по нервам.
Для подтверждения диагноза ЭМГ и мышечная биопсия обычно не требуются, однако эти методы обследования выявляют признаки многочисленных циклов денервации и реиннервации. Активность КФК в крови соответствует норме. В СМЖ возможно повышение уровня белка, цитоз отсутствует.
Разработан точный метод молекулярно-генетической диагностики по анализу крови.
Лечение болезни Шарко-Мари-Тута у детей. Основной проблемой является стабилизация голеностопных суставов. На ранних стадиях заболевания часто помогает ношение жесткой обуви высотой до середины голени, особенно когда пациент ходит по неровной поверхности, например по льду, снегу или по камням. В дальнейшем по мере развития слабости мышц, производящих тыльное сгибание стопы, возможно применение легких пластиковых шин для фиксации задней части голеностопного сустава и нижней части стопы. Они надеваются под носки и невидимы, что особенно удобно для пациентов. При развитии симптома свисающей стопы может потребоваться применение наружных фиксирующих ортопедических приспособлений в области нижней части ноги. В некоторых случаях рассматривается возможность хирургического укрепления голеностопного сустава.
Необходимо защищать ноги от травматического повреждения. В развернутой стадии заболевания можно предотвратить развитие компрессионной невропатии, подкладывая мягкие подушки под голени или между ними во время сна. Парестезия в виде жжения в стопах встречается редко, ее часто купируют приемом фенитоина и карбамазепина. Медикаментозное лечение, способное остановить или замедлить прогрессирование заболевания, не разработано.
Перонеальная мышечная атрофия - аксональный тип. По клинической картине это заболевание напоминает НМСН типа I, однако отличается меньшей скоростью прогрессирования и выраженностью двигательных нарушений. На ЭМГ денервация мышц. Биопсия икроножного нерва выявляет в большей степени аксональную дегенерацию, чем демиелинизацию и луковичные разрастания из шванновских клеток, характерные для НМСН типа I. Аномальный ген локализован на хромосоме 1 в локусе 1р35-р36. Это заболевание отличается от НМСН типа I, хотя оба заболевания наследуются по аутосомно-доминантному типу.
Невральная амиотрофия Шарко-Мари-Тута — это хроническое наследственное заболевание, главным симптомом которого является прогрессирующая мышечная атрофия, локализующаяся в дистальных отделах конечностей, начинающаяся большей частью с нижних, распространяющаяся затем на верхние конечности и в большинстве случаев щадящая черепные нервы и мускулатуру туловища.
Этиология невральной амиотрофии сводится к действию наследственного доминантного фактора; в связи с этим здесь встречается чаще всего непосредственная передача болезни от родителей к детям. Известны случаи, когда болезнь передавалась через 8 поколений. Мужчины заболевают в 1,5 раза чаще женщин. Болезнь распространена по всему земному шару.
Симптомы и признаки болезни
Заболевание развивается постепенно, чаще всего начинаясь в молодом возрасте, но иногда и в раннем детстве. В редких случаях в более позднем возрасте (после 40-ка — 50-ти лет и даже позже). Первые признаки болезни состоят в постепенно нарастающей атрофии мышц нижних конечностей. Атрофии локализуются в дистальных отделах, при этом наблюдается прогрессирующее похудание голеней.
Распределение атрофий может быть различным. Чаще всего поражаются группа разгибателей стопы и пальцев и перонеальная мускулатура, но в дальнейшем процесс может захватить и другие мышечные группы голеней, приводя, в конце концов, к полному параличу стоп (болтающаяся стопа).
По истечении определённого промежутка времени (от одного года до нескольких десятков лет) аналогичный процесс начинает развиваться и в верхних конечностях. Уплощаются возвышение большого пальца и возвышение милинца, западает область отводящей мышцы, область межкостной мускулатуры, рука принимает форму обезьяньей или когтистой лапы, параллельно с атрофией идут нарастающие парезы; ретракций и здесь как правило не образуется. Процесс и здесь медленно распространяется в центральном направлении, захватывая мышцы предплечья, но проксимальные отделы рук и плечевой пояс остаются свободными.
Атрофии при невральной амиотрофии Шарко-Мари-Тута как правило щадят мускулатуру туловища и черепные нервы. Функциональная способность поражённых конечностей может сохраняться парадоксально долгое время. Эти параличи носят все признаки дегенеративных, атрофических параличей. В пораженных мышцах обнаруживается частичная или полная реакция перерождения, часты фибриллярные подергивания. Сухожильные рефлексы угасают, причем нередко это угасание значительно предшествует атрофии и может быть обнаружено и в тех мышечных группах, которые и вовсе не парализуются в дальнейшем течении. Спастические симптомы в чистых случаях отсутствуют. Процесс обычно строго симметричен, хотя по времени одна конечность может быть поражена задолго до появления аналогичного процесса в противоположной конечности.
Прогрессивное распространение атрофий может подвергаться в некоторых случаях модификации такого рода, что верхние конечности заболевают одновременно с нижними, а иногда атрофия их даже предшествует атрофии нижних конечностей. Это начало с рук более характерно для поздно начинающихся случаев болезни Шарко-Мари-Тута.
Наряду с этими характерными моторными симптомами в клиническую картину невротической амиотрофии входят и типичные изменения чувствительности. Сюда относятся прежде всего боли, которые наблюдаются в ряде случаев. Иногда они начинаются задолго до появления атрофий и ослабевают или даже вовсе исчезают в дальнейшем. Боли носят режущий, рвущий характер, локализуются в поражённых конечностях, нередко появляются в виде отдельных приступов, разделенных свободными интервалами, нередко усиливаются после утомления.
Кроме болей могут наблюдаться разнообразные парестезии. При объективном исследовании наблюдается притупление всех видов кожной чувствительности, часто доходящее до степени полной анестезии, без резких границ усиливающееся к дистальным отделам. Периферические нервы могут быть болезненны к давлению. Часто наблюдается болезненный тонический спазм. Пораженные конечности нередко обнаруживают интенсивные вазомоторные расстройства в виде цианоза, похолодания кожи и т. д.
Патологическая анатомия
Патологическая анатомия невральной амиотрофии Шарко-Мари-Тута сводится к комбинации дегенеративных изменений в спинном мозгу и в периферических нервах. В спинном мозгу поражаются задние столбы и клетки передних рогов. Иногда к этой постоянной находке присоединяются и небольшие склеротические изменения в боковых столбах. Находили дегенеративные изменения также в корешках и в спинномозговых ганглиях. Процесс чисто дегенеративный, не сопровождающийся воспалительными изменениями.
В периферических нервах наблюдается картина дегенеративного неврита, усиливающегося по мере удаления от центра и наиболее сильно развитого в периферических нервных разветвлениях. Соединительная ткань нервных стволов разрастается в большей или меньшей степени. Иногда под микроскопом видна эта интерстициальная гиперплазия и в тех случаях, где макроскопически калибр нерва не представлялся измененным. Иногда этот процесс сопровождается размножением ядер шванновской оболочки. Так создаются постепенные переходы к картине настоящего гипертрофического неврита.
Течение болезни
Течение процесса очень медленное и постепенно прогрессирующее. Больные доживают до старости и даже в этих поздних периодах болезни нередко сохраняют способность передвигаться с палкой и в определенной степени пользоваться своими руками.
Заболевание часто принимает в дальнейшем стационарное течение. Однако иногда наблюдаются обострения в связи со случайными внешними причинами (острые инфекции), допускающие впоследствии и некоторое обратное развитие.
В некоторых случаях на картину невральной амиотрофии Шарко-Мари-Тута наслаиваются отдельные невритические симптомы.
Диагностика
Спорадические случаи болезни Шарко-Мари-Тута могут иногда создавать большие трудности для диагностики от хронического полиневрита. Симптоматологическое сходство обеих форм может быть существенным. Хронически прогрессивное течение в спорных случаях решает вопрос в пользу невральной амиотрофии.
Лечение
Терапия чисто симптоматическая: антихолинэстеразные препараты, АТФ, повторные переливания одногруппной крови, витамины группы В, периодический отдых, массаж и электризация атрофирующейся мускулатуры и т. д. Ввиду крайне медленного прогрессирования, иногда показаны ортопедические мероприятия на стопе, которые могут надолго улучшить походку.
Заболевшим амиотрофией Шарко-Мари-Тута показано воздержание деторождения, так как риск развития данного заболевания у ребенка будет составлять 50%; здоровые члены семьи, если они перешли возраст, в котором появляются первые симптомы болезни, могут жениться и иметь детей с минимальным риском передать им болезнь.

Спинальные амиотрофии — это генетические заболевания, проявляющиеся мышечной атрофией и обусловленные дегенеративными изменениями спинальных мотонейронов и моторных ядер ствола головного мозга. Общим симптомокомплексом выступают симметричные вялые параличи с атрофиями мышц и фасцикуляциями на фоне интактной чувствительной сферы. Диагностируются спинальные амиотрофии по данным семейного анамнеза, неврологического статуса, ЭФИ нервно-мышечного аппарата, МРТ позвоночника, ДНК-анализа и морфологического исследования мышечного биоптата. Лечение малоэффективно. Прогноз зависит от формы спинальной мышечной атрофии и возраста ее дебюта.

- Причины
- Патогенез
- Классификация
- Симптомы спинальных амиотрофий
- Болезнь Верднига-Гоффмана
- Ювенильная спинальная амиотрофия Кугельберга-Веландера
- Бульбоспинальная амиотрофия Кеннеди
- Дистальная СМА Дюшенна-Арана
- Скапуло-перонеальная СМА Вюльпиана
- Осложнения
- Диагностика
- Дифференциальная диагностика
- Лечение спинальных амиотрофий
- Немедикаментозная терапия
- Медикаментозная терапия
- Хирургическое лечение
- Экспериментальное лечение
- Прогноз
- Профилактика
- Цены на лечение
Общие сведения
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. Описаны в конце XIX века. Их частота составляет 1 случай на 6-10 тыс. новорожденных. Около 85% спинальных мышечных атрофий составляют проксимальные формы с более выраженной слабостью и атрофиями проксимальных мышечных групп конечностей. На долю дистальных форм приходится лишь 10% СМА. На сегодняшний день спинальные амиотрофии представляют практический интерес для целого ряда дисциплин: детской и взрослой неврологии, педиатрии, генетики.
Причины
Благодаря современной генетике установлено, что возникающие дегенеративные процессы двигательных нейронов обусловлены мутациями в генах SMN, NAIP, H4F5, ВTF2p44, расположенных на 5-ой хромосоме в локусе 5q13. Несмотря на то, что спинальные амиотрофии детерминируются аберрациями одного хромосомного локуса, они представляют собой группу разнородных нозологий, одни из которых проявляются в младенческом возрасте, а другие манифестируют у взрослых. В большинстве случаев амиотрофии наследуются аутосомно-рецессивно.
Патогенез
Генетические мутации приводят к развитию дегенеративных изменений в передних рогах спинного мозга. Нарушается иннервация и нейротрофика поперечно-полосатой мускулатуры. В результате постепенно возникает атрофия мышечной ткани. Преимущественное поражение отдельных групп мышц (проксимальных или дистальных частей верхних или нижних конечностей) у различных форм спинальных амиотрофий отличается. Характерно отсутствие нарушений чувствительности.
Классификация
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
- амиотрофией Верднига-Гоффманна;
- ювенильной формой Кугельберга-Веландера;
- хронической инфантильной СМА;
- синдромом Виалетто-ван Лэре (бульбоспинальная форма с глухотой);
- синдромом Фацио-Лонде.
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
- бульбоспинальная амиотрофия Кеннеди;
- скапулоперонеальная;
- лицелопаточноплечевая и окулофарингеальная формы;
- дистальная СМА;
- мономелическая СМА.
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Симптомы спинальных амиотрофий
Общим для спинальных мышечных атрофий является симптомокомплекс симметричного вялого периферического паралича: слабость, атрофия и гипотония мышечных групп одноименных конечностей (чаще вначале обеих ног, а затем и рук) и туловища. Пирамидные нарушения не типичны, но могут развиваться на поздних стадиях. Расстройства чувствительности отсутствуют, функция тазовых органов сохранена. Обращает внимание более выраженное поражение проксимальных (при проксимальных СМА) или дистальных (при дистальных СМА) мышечных групп. Типично наличие фасцикулярных подергиваний и фибрилляций.
Встречается в 3-х клинических вариантах. Врожденный вариант дебютирует в первые 6 мес. жизни и является наиболее злокачественным. Его симптомы могут проявляться еще во внутриутробном периоде слабым шевелением плода. Дети с рождения имеют мышечную гипотонию, не способны переворачиваться и держать голову, при более позднем дебюте — не могут сидеть. Патогномонична поза лягушки — ребенок лежит с разведенными в стороны и согнутыми в коленях и локтях конечностями.
Амиотрофии имеют восходящий характер — вначале возникают в ногах, затем вовлекаются руки, позже — дыхательная мускулатура, мышцы глотки и гортани. Сопровождается задержкой психического развития. К 1,5 годам наступает смертельный исход.
Ранняя спинальная амиотрофия манифестирует до 1,5 лет зачастую после инфекционного заболевания. Ребенок утрачивает двигательные способности, не может стоять и даже сидеть. Периферические парезы сочетаются с контрактурами. После вовлечения дыхательных мышц развивается дыхательная недостаточность и застойная пневмония. Летальный исход обычно происходит в возрасте до 5-ти лет. Поздний вариант дебютирует после 1,5 лет, отличается сохранением двигательной способности до 10-летнего возраста. Летальный исход наступает к 15-18 годам.
Характеризуется дебютом в период от 2 до 15 лет. Начинается с поражения проксимальных мышц ног и тазового пояса, затем захватывает плечевой пояс. Около четверти пациентов имеют псевдогипертрофии, что делает клинику сходной с проявлениями мышечной дистрофии Беккера. В плане дифдиагностики большое значение имеет наличие мышечных фасцикуляций и данные ЭМГ. Течение амиотрофии Кугельберга-Веландера доброкачественное без костных деформаций, в течение ряда лет пациенты остаются способными к самообслуживанию.
Наследуется рецессивно сцеплено с Х-хромосомой, манифестирует только у мужчин после 30-летнего возраста. Типично медленное, относительно доброкачественное течение. Дебютирует с амиотрофии проксимальных мышц ног. Бульбарные расстройства появляются через 10-20 лет и благодаря медленному прогрессированию не вызывают нарушения витальных функций. Может наблюдаться тремор головы и рук. Патогномоничным симптомом выступают фасцикулярные подергивания в периоральных мышцах. Зачастую отмечается эндокринная патология: атрофия яичек, снижение либидо, гинекомастия, сахарный диабет.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем – эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Диагностика
Больных со спинальными амиотрофиями курируют врачи-неврологи, а при манифестации в детском возрасте – педиатры или неонатологи. При необходимости может потребоваться привлечение для консультации других специалистов – эндокринологов и генетиков. Для диагностики некоторых разновидностей амиотрофий большое значение имеют анамнестические данные, а именно возраст появления симптоматики (например, болезнь Верднига-Гоффмана всегда дебютирует у детей до 6 месяцев, а амиотрофия Кугельберга-Веландера – после 2 лет).
Во время осмотра пациента обращается внимание на снижение общего мышечного тонуса, ослабление или утрату сухожильных рефлексов, скелетно-мышечные деформации. У части больных отмечается псевдогипертрофия икроножных мышц. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
Дифференциальный диагноз спинальных амиотрофий необходимо проводить с другими наследственными нейродегенеративными заболеваниями, поражающими мышечную ткань. К таким патологиям можно отнести:
Лечение спинальных амиотрофий
Все без исключения больные должны быть госпитализированы в стационар. В тяжелых ситуациях (например, при дыхательной недостаточности вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. Все мероприятия направлены на облегчение состояния пациента. Несмотря на это, комплексный подход и строгое соблюдение рекомендаций врачей позволяют улучшить качество жизни больного. Виды консервативной терапии, применяющиеся для терапии спинальных амиотрофий:
- Обеспечение питания. При значительном нарушении акта глотания особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение пациента вертикальным. Может понадобиться установка назогастрального зонда.
- ЛФК. С целью повышения тонуса мышц и замедления их атрофии рекомендуются регулярные физические нагрузки. Наиболее эффективно совмещение активных (выполняются специалистом) и пассивных упражнений (выполняются самим пациентом).
- Физиолечение. Для активации метаболизма в мышечных тканях назначаются сеансы электростимуляции модулированным током, грязевые аппликации, электрофорез.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Ортопедическое лечение. Для предупреждения и коррекции костных деформаций и суставных контрактур рекомендуется использование ортопедических приспособлений: корсетов, ортезов, ортопедической обуви.
- Респираторная поддержка. Нередко возникает необходимость в устранении кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно проводиться непрерывно и подбираться индивидуально для конкретного пациента.
Этиотропная терапия с доказанной эффективностью на сегодняшний день отсутствует. Лекарственные препараты, применяющие для лечения спинальных амиотрофий, следующие:
- Метаболические средства. Для улучшения обменных процессов в мотонейронах и мышечных тканях применяются препараты, стимулирующие функцию митохондрий (коэнзим Q10, янтарная кислота), ноотропы (пирацетам, гамма-аминомасляная кислота), L-карнитин.
- Вальпроаты и Кленбутерол. Исследования показали, что противоэпилептические препараты из группы производных вальпроевой кислоты и агонист бета-адренергических рецепторов Кленбутерол способны увеличивать образование белка выживаемости мотонейронов (SMN) и, соответственно, улучшать клиническое течение заболевания.
- ИПП и прокинетики. Пациентам с гастроэзофагальным рефлюксом, который нередко развивается при нарушении глотания, назначаются ингибиторы протонной помпы (эзомепразол) и средства, стимулирующие моторику желудочно-кишечного тракта (итоприд).
- Муколитики и отхаркивающие средства. У больных со слабостью дыхательной мускулатуры для устранения таких проблем как слабое отхаркивание и скопление в дыхательных путях густой мокроты применяются препараты, разжижающие мокроту (ацетилцисетин) и стимулирующие ее отхаркивание (терпингидрат).
- Гормональные препараты. Людям с возникшими эндокринологическими осложнениями показаны инсулин, сахароснижающие препараты (метформин), тестостерон и антиандрогены (дутастерид).
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. Лежачим больным, страдающим постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Постоянно ведутся многочисленные исследовательские работы по разработке лекарственных средств, способных остановить или хотя бы замедлить процесс нейродегенерации. Значительные успехи достигнуты в области генной терапии. Уже используются в клинической практике препараты, корректирующие дефекты матричной РНК гена SMN – Спинраза и Рисдиплам. В конце 2019 года был зарегистрирован и одобрен к клиническому применению препарат Золгенсма, который содержит функционально полноценный ген SMN1.
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Профилактика
Специфических методов первичной профилактики не существует. Единственный способ предотвратить возникновение болезни – пренатальная диагностика (обнаружение мутаций в ворсинах хориона или амниотической жидкости) с прерыванием беременности. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Читайте также: