
Склеродермия очаговая дифференциальная диагностика
За последнее десятилетие существенно расширились представления о системных заболеваниях соединительной ткани, среди которых второе место по частоте занимает склеродермия.
За последнее десятилетие существенно расширились представления о системных заболеваниях соединительной ткани, среди которых второе место по частоте занимает склеродермия. Заболевание характеризуется системным прогрессирующим поражением соединительной ткани с преобладанием фиброзно-склеротических и сосудистых изменений по типу облитерирующего эндартериита с распространенными вазоспастическими расстройствами [7, 12].
Несмотря на отсутствие официальных статистических данных, можно утверждать, что больных с таким аутоиммунным заболеванием, как очаговая склеродермия, становится все больше и протекает это заболевание агрессивнее [15]. Возможно, это связано с несоблюдением норм диспансеризации и сроков лечения [11].
ОСД, так же как и ССД, чаще болеют лица женского пола, например, девочки болеют чаще мальчиков более чем в 3 раза, а женщины в возрасте 40–55 лет составляют 75% больных склеродермией [21]. Заболевание может возникать в любом возрасте, даже у новорожденных, начинаясь обычно без каких-либо субъективных ощущений и нарушения общего состояния. В связи с тенденцией растущего организма к распространению патологии, к выраженным сосудистым реакциям у детей это заболевание часто имеет склонность к обширному поражению, хотя в ранние сроки может проявляться единичными очагами.
Патогенез склеродермии связывают главным образом с гипотезами обменных, сосудистых и иммунных нарушений. На возникновение ОСД влияют также нарушения вегетативной нервной системы и нейроэндокринные расстройства. Принято рассматривать ограниченную склеродермию как своеобразное аутоиммунное заболевание, в основе которого лежат аутоиммунные и воспалительные реакции на различные антигены. В. А. Владимирцев и соавт. (1982) считают, что повышенный уровень коллагеновых белков, являясь источником активной антигенной стимуляции, создает фон, на котором при генетической предрасположенности реализуются аутоиммунные реакции. Возникающий порочный круг взаимовлияния лимфоидных и коллагенсинтезирующих клеток ведет к прогрессированию фиброзного процесса [6]. Установленные нарушения гуморального и клеточного иммунитета у больных склеродермией чаще регистрировались у женщин. Клеточный иммунитет у женщин, в отличие от его гуморального звена, менее активен по сравнению с мужским. Снижение клеточного иммунитета, особенно его супрессорного звена, при повышении активности гуморального иммунитета приводит к тому, что у женщин гораздо чаще, чем у мужчин, развивается аутоиммунный процесс. Прослеживается связь склеродермии с беременностью и менопаузой [21]. В последние годы появились исследования об участии эстрогенов и прогестерона, а также некоторых других гормонов в реакциях синтеза коллагена и других компонентов соединительной ткани. Особое патогенетическое значение при склеродермии придают изменениям микроциркуляции, которые наиболее выражены в период менопаузы. В их основе лежат поражения преимущественно стенок мелких артерий, артериол и капилляров, пролиферация и деструкция эндотелия, гиперплазия интимы [3, 5, 12, 16, 20]. До сих пор обсуждается вопрос о роли наследственности в развитии ОСД. По данным Furst A. (2004) коренные индейцы штата Оклахома в 8 раз чаще болеют склеродермией, чем другие жители Соединенных Штатов. Также более подвержены данной болезни чернокожие люди, они чаще заболевают в детском возрасте и имеют более распространенный процесс по сравнению с белокожими. Однако исследования, проведенные тем же автором, установили, что всего 6% близнецов одновременно болеют склеродермией, и это недостаточно высокий процент заболеваемости среди близнецов, чтобы утверждать чисто генетическую этиологию болезни.
Противоречия данных литературы, вероятно, обусловлены тем, что характер и выраженность иммунологических, эндокринных и обменных сдвигов в значительной степени зависят от течения заболевания в целом и от степени поражения индивидуально [8].
До настоящего времени многие исследователи продолжают поддерживать инфекционную теорию возникновения склеродермии. Развитие склеродермии может быть связано с перенесением таких заболеваний, как грипп, ангина, скарлатина, пневмония. Отдельные авторы рассматривают распространенную склеродермию как позднее проявление боррелиоза (син.: иксодовый клещевой боррелиоз, Лайма болезнь), что подтверждается определением у некоторых больных (особенно бляшечной и склероатрофическими формами) высокого титра иммуноглобулиновых антител к боррелиям Бургдорфера и поразительно быстрым улучшением после лечения заболевания пенициллином. S. Вucher, основываясь на результатах иммунологических исследований и обнаруженных в замороженных биоптатах спирохетоподобных структур, посчитал это подтверждением спирохетной теории возникновения склеродермии [9, 18]. Проведенные наблюдения установили различные кожные проявления Лайм-боррелиоза: бляшечная форма склеродермии (98%), атрофодермия Пазини–Пьерини (80%), анетодермия и хронический атрофический акродерматит (100%) и редко — склероатрофический лихен [4, 9, 16, 18].
В противовес приведенным данным многие исследователи склонны расценивать случаи ограниченной склеродермии с большим титром антител к боррелиям и выявление спирохет — как боррелиоз, протекающий под маской ограниченной склеродермии, а склерозирование кожи — как псевдосклеротические изменения, но ни в коем случае не как проявления истинной склеродермии. По мнению Н. С. Потекаева и соавт. (2006), патогенетическая связь ОСД с болезнью Лайма, также как и атрофодермии Пазини–Перини, синдрома Перри–Ромберга лишь предполагается [17]. Для подтверждения наличия болезни Лайма у больного со склеродермическими очагами целесообразно определение специфических антител в сыворотках больных методами непрямой реакции иммунофлюоресценции (НРИФ), полимеразной цепной реакции (ПЦР), а также выявление боррелий в биоптатах кожи из очагов поражения методом серебрения [9, 18].
Несмотря на разнообразие теорий возникновения ОСД, ни одна из них не раскрывает инициальную причину и взаимодействие факторов патогенеза склеродермического процесса. Наиболее интересными представляются исследования некоторых показателей кальциевого обмена, проведенных Болотной Л. А. и соавт. (2004). Авторы на основании полученных результатов сделали вывод, что изменения кальция и магния, выявленные на всех этапах ОСД, имеют патогенетическое значение. Степень этих расстройств находится в прямой зависимости от активности, формы и длительности дерматоза. Дефект функций клеточных мембран может обуславливать накопление кальция в разных клетках больных ОСД и усиливать синтетическую активность фибробластов, сужение сосудов микроциркуляторного русла, стимуляцию лимфоцитов. Гипомагниемия, выявленная у данных больных, способствует дестабилизации клеточных мембран и может быть одной из причин накопления кальция в эритроцитах, а также обуславливать нарушение функции ряда ферментов [4].
Единой общепринятой классификации ОСД не существует, на наш взгляд, более приемлема классификация С. И. Довжанского (1979), в которой наиболее полно представлены все клинические формы ОСД:
Более редкой разновидностью ограниченной склеродермии является полосовидная (линейная), наблюдаемая обычно у детей. Отличие от бляшечной склеродермии заключается только в очертаниях очагов — они имеют вид полос и располагаются обычно на конечностях и по саггитальной линии на лбу (напоминают рубец от удара саблей).
Другая разновидность склеродермии — лихен склероатрофический (склеродермия каплевидная, болезнь белых пятен, лишай белый Цумбуша). Предполагают, что он может являться атрофической формой красного плоского лишая, или крауроза; не исключается самостоятельность дерматоза. Однако часто наблюдается сочетание бляшечной склеродермии и склероатрофического лихена, что говорит о единстве этих клинических форм. Высыпания при склероатрофическом лихене представлены мелкими рассеянными или сгруппированными белесоватыми пятнами, иногда с ливидным оттенком, размерами 0,5–1,5 см, чаще локализующиеся на коже туловища и шее, а также на любом участке кожного покрова. Нередко заболевание развивается у девочек и молодых женщин в области половых органов. Встречаются распространенные формы склероатрофического лихена и атипичные варианты; буллезная и телеангиоэктатическая [2, 10, 19]. Для буллезной формы характерно образование пузырей с плотной покрышкой и серозным содержимом. Пузыри могут вскрываться, обнажая эрозии, или ссыхаться в плотную серозную корку. Пузыри свидетельствуют о прогрессировании атрофического процесса, и если на их месте впоследствии образуются эрозии и язвы, процесс трудно поддается терапии. При телеангиоэктатической форме на участках белесоватой атрофии кожи образуются телеангиоэктазии [12].
Склероатрофический лихен вульвы (САЛВ) считается редким заболеванием, однако у детей заболевание встречается не столь редко, как это следует из данных зарубежной литературы. Большинство детей (70%) заболевает в возрасте до 10–11 лет, т. е. до начала пубертатного периода. САЛВ считается заболеванием с неизвестной этиологией и патогенезом, некоторые авторы отмечают участие гормонального фактора в его патогенезе. В частности, Е. А. Бурова (1989) указывает на ведущую роль дисгормональных нарушений в системе гипофиз — надпочечники — яичники. Клиническая картина САЛВ представлена образованием небольших склероатрофических очагов беловато-сероватого цвета, иногда с перламутровым оттенком, блеском, точечными углублениями, фолликулярным кератозом, сиреневым краем. Атрофические изменения наиболее выражены при локализации в области вульвы. Для девочек с САЛВ в связи с низким уровнем эстрогенов характерны более поздние сроки полового созревания, менструальная дисфункция [2, 5, 10, 19].
Атрофодермия Пазини–Пьерини характеризуется немногочисленными пятнами, которые располагаются преимущественно на спине и имеют, как правило, большие размеры (до 10 см и более) и часто неправильные очертания. Заболевание является как бы переходной формой между бляшечной склеродермией и атрофией кожи. Эта разновидность обычно наблюдается у молодых женщин. Высыпания — в виде синевато-фиолетовых пятен с гладким, слегка западающим центром, но без феномена проваливания пальца или грыжевидного выпячивания. Иногда вокруг пятна видно сиреневое кольцо. Характерным признаком этой формы ОСД является длительное отсутствие уплотнения в начале заболевания. В ряде случаев отчетливо выражена пигментация. Одновременно с клиническими проявлениями атрофодермии Пазини–Пьерини могут наблюдаться типичные проявления ОСД [2, 10, 19]. Хотя высказывается мнение, что атрофодермия является самостоятельным заболеванием, все же, по-видимому, правильнее рассматривать ее как клиническую разновидность ОСД, тем более что в ряде случаев атрофия и гиперпигментация предшествуют развитию склероза, который все же появляется на бляшках атрофодермии лишь через несколько лет. Отличие идиопатической атрофодермии от бляшечной склеродермии состоит в том, что при атрофодермии поражается главным образом кожа туловища, а не лица и конечностей, а сам процесс развивается длительно (в течение нескольких лет), очаги поражения представляют собой бляшки почти без уплотнения, синевато-коричневого цвета без лилового кольца по периферии. Полного регресса атрофодермии не наблюдается, в то время как очаг бляшечной склеродермии может исчезнуть полностью (при вовремя начатом лечении) или после него остается легкая атрофия или стойкая пигментация [2, 10, 19].
Одним из редких проявлений склеродермии является гемиатрофия лица Парри–Ромберга — заболевание, характеризующееся прогрессирующей атрофией только одной половины лица, проявляющееся дистрофическими изменениями кожи и подкожной клетчатки, в меньшей степени — мышц и лицевого скелета. Общее состояние больных, как правило, остается удовлетворительным, главной жалобой является косметический дефект в области лица. По данным литературы, среди больных преобладают женщины. В большинстве наблюдений заболевание развивается в возрасте от 3 до 17 лет. Одинаково часто отмечается левосторонняя и правосторонняя локализация процесса. Как правило, заболевание имеет длительное, хроническое течение. Активная стадия длится в основном до 20 лет, в некоторых наблюдениях — до 40 лет. Первыми признаками заболевания являются локальные изменения кожи лица, которая вскоре приобретает желтоватый или синюшный оттенок. Постепенно развивается уплотнение кожи в очагах. В дальнейшем в местах уплотнения кожа атрофируется; с течением времени атрофические изменения прогрессируют с вовлечением в процесс подкожной жировой клетчатки и мышц лица. Наиболее выраженными и частыми признаками поражения кожи являются резкое ее истончение, морщинистость, гиперпигментация диффузного или очагового характера. В атрофированных участках кожи отсутствует рост волос. У больных страдает не только кожа, но и подлежащие мягкие ткани, что, как правило, приводит к грубейшей деформации лица в виде значительной асимметрии его правой и левой половины, наиболее выраженной при дебюте заболевания в раннем детском возрасте. Костные структуры тоже поражаются, если гемиатрофия возникает до окончания их роста. У части больных наблюдается атрофия половины языка. Имеются клинические наблюдения развития прогрессирующей гемиатрофии лица как проявление терминальной стадии у больных с агрессивным течением полосовидной склеродермии на лице. В литературе приводятся данные о результатах обследования больных склеродермией, у 16,7% из которых в последующем развилась лицевая гемиатрофия. Такие случаи дают основание предполагать, что гемиатрофия Ромберга является неблагоприятным вариантом течения ОСД [1].
Диагностика ограниченной склеродермии может представлять определенные трудности, особенно в начальной стадии заболевания. Дифференциальный диагноз ОСД проводят с витилиго, краурозом, недифференцированной формой лепры, синдромом Шульмана.
В начале развития бляшечной склеродермии, когда уплотнение еще не выражено и имеется только обесцвеченное пятно, процесс может напоминать витилиго или депигментированное пятно при недифференцированной лепре. При витилиго пятна имеют более четкую границу, которая хорошо видна при наличии гиперпигментированной зоны. Поверхность пятен гладкая, без признаков атрофии и шелушения. Пятна витилиго сохраняются довольно длительно без уплотнения.
При недифференцированной лепре изменения на коже характеризуются пятнистыми высыпаниями. Последние могут быть эритематозными, различных оттенков (от розового до синюшного) и гипопигментированными. В области пятен болевая, тактильная и температурная чувствительность снижена.
Труднее дифференцировать линейную склеродермию от линейно расположенного келлоидоподобного невуса. Отличительным признаком может служить обнаружение келлоидоподобного невуса в первые месяцы жизни и его длительное существование без выраженных изменений на протяжении многих лет.
Крауроз вульвы может в известной мере напоминать склеродермию, в частности склероатрофический лихен, поскольку при этом заболевании поверхность пораженных участков сухая, блестящая, плотная. Однако при краурозе вульвы имеются интенсивный зуд и телеангиоэктазии. В дальнейшем развиваются атрофия малых и больших половых губ, лейкоплакия и, нередко, рак. Крауроз полового члена проявляется в виде хронической атрофии и сморщивания головки полового члена и внутреннего листка крайней плоти, тогда как склеротические изменения крайней плоти и головки полового члена вызывают фимоз и сужение отверстия мочеиспускательного канала. В противоположность краурозу вульвы, при поражениях полового члена зуд отсутствует и болезнь не осложняется раком.
Синдром Шульмана (син.: эозинофильный фасциит, диффузный фасциит с гипергаммаглобулинемией и эозинофилией). Под этим заболеванием понимают диффузное склеродермоподобное уплотнение кожи, утолщение мышечной фасции, инфильтрацию ее эозинофилами, лимфоцитами и плазматическими клетками. Очаги локализуются чаще на конечностях и приводят к сгибательным контрактурам. Дифференциальными признаками, характерными для синдрома Шульмана, служат отсутствие фиолетового венчика вокруг очага уплотнения и атрофии кожи, а также наличие болевого синдрома и эозинофилия в периферической крови [2, 10, 19].
При локализации очага склеродермии на лице нужно помнить о такой редкой разновидности опухоли, как склеродермоподобная форма базалиомы, при которой патогномоничный для базалиомы узелок медленно увеличивается в размерах, трансформируется в плотную, слегка возвышающуюся над поверхностью кожи бляшку цвета слоновой кости с восковидным блеском, в центральной части которой видны телеангиоэктазии. Границы очага резкие, очертания округлые или неправильные, размеры от 1 до 3 см и более. Эта форма представляет трудность для диагностики, если игнорировать имеющиеся по периферии очага узелки, патогномоничные для базалиомы.
Лечение. Терапия ОСД должна быть многокурсовой и многокомпонентной. При активном процессе количество курсов должно быть не менее 6, с интервалом 1–2 месяца; если процесс стабилизировался, интервал между курсовым лечением увеличивается до 4 месяцев; при остаточных клинических проявлениях и в целях профилактики проводится терапия 2–3 раза в год препаратами, улучшающими микроциркуляцию.
При активном процессе в лечение ОСД следует включать следующие группы препаратов:
При единичных очагах поражения можно ограничиться назначением витамина В12 в свечах и фонофореза с Лидазой, Ронидазой, трипсином, хемотрипсином (№ 7–10). Местное лечение ОСД должно состоять из аппликаций наружных средств и физиотерапии. В топической терапии ОСД обычно используются следующие мази: Гепариновая, Гепароид, Троксовазиновая, Бутадионовая, Теониколовая. Препаратами выбора топической терапии являются Димексид, Унитиол, Ронидаза, трипсин, химотрипсин, Лидаза, которые могут применяться в виде аппликаций или вводиться в очаги поражения с помощью электро- и фонофореза. Ронидазу применяют наружно, нанося ее порошок (0,5–1,0 г) на смоченную физиологическим раствором салфетку. Накладывают салфетку на очаг поражения, фиксируя бинтом в течение полусуток. Курс аппликаций продолжают 2–3 нед. Рекомендуется назначать ультрафонофорез Купренила и Гидрокортизона на очаги поражения. При ОСД применяют также магнитотерапию, вакуум-декомпрессию, низкоинтенсивную лазеротерапию. В конце курса терапии можно присоединить массаж очагов поражения. При спаде активности процесса — сероводородные и родоновые ванны [10, 19, 20].
В последние годы широкое применение при лечении различной патологии получила гипербарическая оксигенация (ГБО), способствующая более интенсивному обогащению тканей кислородом, который при повышенном давлении может оказывать также противомикробное действие, особенно в отношении анаэробных микроорганизмов. ГБО увеличивает метаболическую активность митохондрий и их способность к регенерации, нормализует окисление липидов, повышает уровень утилизации кислорода тканями в связи с активацией аэробных процессов в очагах поражения, улучшает микроциркуляцию. На целесообразность использования ГБО при склеродермии указывают ряд авторов [3, 13].
Многие авторы рекомендуют плазмозамещающие препараты декстрана (Декстран, Реомакродекс) [3, 10, 16]. Однако, на наш взгляд, к такой терапии следует прибегать при распространенных, быстропрогрессирующих формах ОСД.
Нами не отмечено значительного эффекта от лечения гормонами, анаболиками и препаратами хинолинового ряда.
В заключение хочется подчеркнуть, что к лечению больных ОСД нужно подходить индивидуально в зависимости от стадии процесса, распространенности, наличия сопутствующих заболеваний. Необходимо объяснить больному целесообразность длительной терапии, тщательного обследования и профилактического лечения.
Очаговая склеродермия – это хроническое прогрессирующее аутоиммунное заболевание соединительной ткани, характеризующееся появлением на различных участках тела очагов локального воспаления (эритемы и отека), с последующим формированием в них склероза и/или атрофии кожи и подкожной жировой клетчатки. Данная патология встречается у представителей любой расы. Женщины болеют чаще, чем мужчины (2,7:1). Заболевание может возникать в любом возрасте, даже у новорожденных, начинаясь без какой-либо симптоматики и ухудшения общего состояния. Согласно эпидемиологическим данным, очаговая склеродермия (см. фото №1) занимает второе место по распространённости среди аутоиммунных заболеваний соединительной ткани. За последнее десятилетие увеличилась тенденция к более агрессивному течению данной патологии, что, возможно, связано с несоблюдением норм диспансеризации и сроков лечения.
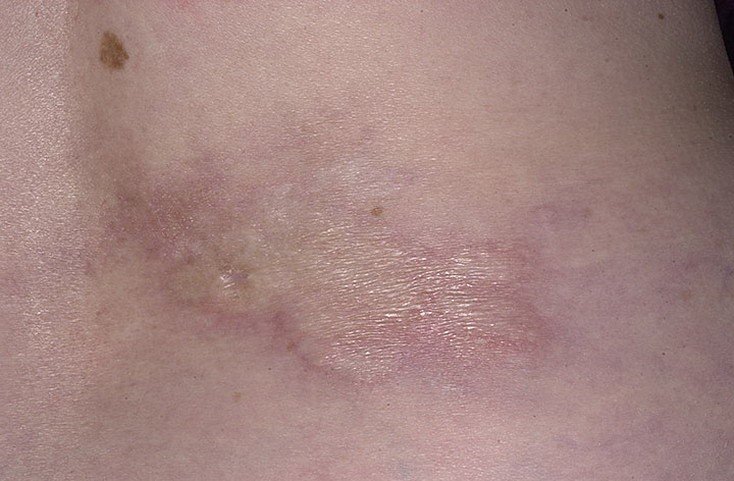
Очаговая склеродермия(фото№1)
У больных со склеродермией всегда есть риск перехода локализированного процесса в системный. Выделяют четыре основных прогностические фактора, которые говорят о вероятности диффузного течения данного заболевания, а именно:
- Дебют заболевания в возрасте до 20 лет или после 50 лет;
- Множественная бляшечная или линейная формы заболевания;
- Локализация очагов поражения над суставами конечностей;
- Дисиммуноглобулинемия.
В Юсуповской больницы диагностикой и лечением различных аутоиммунных заболеваний соединительной системы занимаются врачи-ревматологи. Для своевременного обследования и проведения дальнейшего лечения клиника оснащена мощной диагностической базой, которая представлена целым рядом инновационного медицинского оборудования европейского образца. В дневном стационаре клиники пациент сможет пройти быстро и комфортно все необходимые процедуры, минуя часовые стояния в очереди.
Очаговая склеродермия: этиология и патогенез
В большинстве случаев причины возникновения очаговой склеродермии остаются неизвестные. В патогенезе заболевания основную роль играют аутоиммунные нарушения, повышение синтеза и отложение в коже и подкожной жировой клетчатке коллагена и других компонентов соединительной ткани, микроциркуляторными расстройствами. Шифр по очаговой склеродермии по Международной классификации болезней МКБ-10соответствует L94.0
Очаговая склеродермия: классификация
На данном этапе общепринятой классификации не разработано. Наиболее приемлема классификация С.И. Довжанского (1979) очаговой склеродермии (см. фото № 2) у взрослых, которая включает в себя все клинические формы:
- Бляшечная склеродермия; очаговая, узловатая;
- Линейная склеродермия;
- Генерализованная склеродермия
- Глубокая склеродермия;
- Пансклеротическая склеродермия;
- Буллезная склеродермия;
- Идиопатическая атрофодермия Пазини-Пьерини;
- Прогрессирующая гемиатрофия лица Парри-Ромберга;
- Склероатрофический лихен.

Очаговая склеродермия (фото №2) у взрослого. Линейная форма.
Очаговая склеродермия: клиническая картина
Клиническая картина очаговой склеродермии разнообразна. Больные на приеме у врача-ревматолога могут предъявлять жалобы на зуд, болезненность, чувство покалывания и стянутости кожи, ограничение движения в суставах, деформацию пораженных участков кожи.
Классически принято выделять три стадии развития данной патологии соединительной ткани:
- Эритема (отек) – появление покраснения кожи в области поражения;
- Склероз – появление уплотнения кожи;
- Атрофия кожи – истончение кожи и подкожно-жирового слоя.
Наиболее распространенной формой очаговой склеродермии является бляшечная склеродермия, которая характеризуется появлением на туловище или конечностях очагов эритемы и индурации (уплотнение) кожи. Центр очагов со временем светлеет, приобретает оттенок слоновой кости, и все больше затвердевает. Процесс склерозирования продолжается пока не сформируется блестящий атрофический пласт. В течение месяца он разрешается, приобретая коричневатую гиперпигментацию.
При узловатой склеродермии появляются единичные или множественные узелки, внешне похожие на келоидные рубцы. Как правило, они появляются у больных, не имеющих склонности к развитию келоидов. Кожа в области склеродермии имеет телесный цвет.
При линейной склеродермии на кожи появляются очаги эритемы линейной формы. Характерная локализация очагов по ходу сосудисто-нервного пучка или на одной половине тела.
Очаговая склеродермия: диагностика
Диагностика очаговой склеродермии основывается на данных анамнеза заболевания, анамнеза жизни, жалоб и клинической картине заболевания. Данную патологию следует лечить и наблюдать у врача-ревматолога. Для исключения сопутствующих заболеваний и противопоказаний пациент должен быть проконсультирован следующими специалистами:
- Терапевтом;
- Кардиолог;
- Эндокринологом;
- Гинекологом;
- Невропатологом;
- Гастроэнтерологом;
- Отоларингологом.
Для уточнения активности патологического процесса, а также исключения системной гипертермии назначают проведение следующих исследований:
- Лабораторные обследования: ОАК, ОАМ, биохимический анализ крови;
- Гистологическое исследование кожи;
- Определение в крови антинуклеарного фактора;
- УЗИ органов брюшной полости, почек, щитовидной железы;
- ЭКГ;
- Рентгенография грудной клетки;
- Компьютерная томография;
- Магнитно-резонансная томография.
Очаговая склеродермия: лечение
Лечение очаговой склеродермии в Юсуповской больнице подбирается в индивидуальном порядке каждому пациенту в зависимости от формы, стадии и тяжести течения заболевания, а также локализации очагов поражения. Терапия при данном заболевании соединительной ткани должна быть направлена на:
- Снижение активности патологического процесса и прогрессирование заболевания;
- Уменьшение площади поражения кожи и выраженность клинических симптомов заболевания;
- Предотвращение развития осложнений;
- Улучшение качества жизни больных.
При наличии сгибательных контрактур, деформаций скелета и косметических дефектов кожи необходима консультация хирурга для решения вопроса о проведении хирургической коррекции.
Больные с активным, быстро прогрессирующим течением заболевания показано введение в медикаментозную терапию глюкокортикостероидных препаратов системного действия.
Больным с тяжелыми формами очаговой склеродермии со сформированными глубокими поражениями кожи и подлежащих тканей назначают цитостатические препараты (метотрексат) в виде монотерапии или в комбинации с глюкокортикоидными препаратами системного действия. Иногда может наблюдаться спонтанный регресс склерозирующего процесса кожи или полное разрешение очагов поражения.
В Юсуповской больнице ведущие специалисты с многолетним стажем работы ведут пациентов с различными формами очаговой склеродермии. В стенах стационара пациент окунется в комфортабельную и уютную атмосферу, благодаря профессиональной работе высокоспециализированного медицинского персонала. После курса лечения для каждого больного разрабатывается программа реабилитации. Звоните по телефону для записи на прием к врачу-ревматологу.
Ограниченная склеродермия встречается относительно часто, но преимущественно в дерматологической практике. Клиническая картина болезни в отличие от ССД обычно ограничивается локальным поражением кожи; синдром Рейно и висцеральные проявления, т. е. системность процесса, отсутствуют. Эволюция и прогноз в целом благоприятны, однако нередко наблюдают длительное течение, иногда — сравнительно тяжелые прогрессирующие формы (например, линейная склеродермия у детей с односторонним, но распространенным глубоким поражением тканей и нарушением роста конечностей, приводящим к инвалидизации).
Линейная склеродермия развивается, как правило, у детей и подростков, а очаговая может выявиться в любом возрасте, причем генерализованные формы ее с распространенным, иногда диффузным поражением кожи туловища начинаются иногда в преклимактерический период.
Кроме того, выделяют атипичные варианты очаговой склеродермии, к которым относят подкожную (глубокую) узловатую псевдопойкилодермическую, буллезную и атрофическую формы.
Чаще встречается бляшечная склеродермия, характеризующаяся наличием одного и более очагов различной локализации (туловище, конечности) чаще белого цвета с лиловым ободком в активный период заболевания. В очаге поражения кожа уплотнена и спаяна с подлежащими тканями или атрофична; иногда имеются участки гиперпигментации. Как клинически, так и морфологически патологический процесс в участке поражения сходен с наблюдающимся при ССД. Это локализованный фиброз или склероз кожи, подкожной клетчатки с увеличением коллагеновых волокон и их гомогенизацией, уменьшением придатков кожи, изменением мелких сосудов. На ранней стадии поражения отмечается воспалительная инфильтрация лимфоцитами, плазматическими клетками и макрофагами.
При линейной склеродермии нередко поражаются фасции, мышцы, околосуставные ткани, на ранних этапах выражена воспалительная инфильтрация, которая также свойственна генерализованной очаговой склеродермии.
Хотя системность поражения при этих формах отсутствует, в очагах поражения кожи и подлежащих тканей возможно развитие контрактур, суставной и мышечной патологии, а при склеродермии типа удара саблей в области лица и волосистой части головы — вовлечение в процесс костей черепа, глазная (односторонняя) и неврологическая симптоматика.
Лабораторные сдвиги чаще отсутствуют, но у некоторых больных выявляют антинуклеарные антитела, гипергаммаглобулинемия, повышение уровня циркулирующих иммунных комплексов, что свидетельствует об участии иммунного компонента в патогенезе заболевания.
Больные ограниченными формами склеродермий обычно наблюдаются и лечатся дерматологом, при наличии суставных, сосудистых и висцеральных проявлений консультируются ревматологом и терапевтом, а при выявлении системности процесса подлежат лечению, аналогично больным ССД.
Диффузный эозинофильный фасциит (ДЭФ) выделен в самостоятельную форму на основании преимущественной локализации склеродермоподобных (индуративных) поражений в области предплечий и голеней при сохранности дистальных отделов конечностей, эозинофилии и преобладании патологических изменений в фасциях и прилежащих тканях при морфологическом исследовании [Shuiman Р., 1974; Kahn M., Grossin M., 1985].
Отличается от ССД также отсутствием синдрома Рейно и висцеральной патологии, хотя в последнее время описаны поражение отдельных органов и тяжелые гематологические осложнения (апластическая анемия, тромбоцитопеническая пурпура) иммунного генеза, что возобновило дискуссию о нозологической самостоятельности и благоприятном прогнозе заболевания.
Характерны еще две особенности эозинофильного фасциита: 1) развивается нередко после чрезмерной или необычной для данного индивидуума нагрузки и 2) преимущественно у мужчин, что может быть в некотором смысле и взаимосвязано, но противоположно соотношению полов, в частности, при ССД. Эти особенности могут быть дополнены, как показали собственные наблюдения 23 больных эозинофильным фасциитом и данные литературы, преимущественно острым началом ДЭФ (с развитием индуративных изменений в течение первого месяца, а иногда и недели!) и более ярким, чем при ССД, воспалительным компонентом (выраженная лимфоплазмоклеточная инфильтрация с примесью эозинофилов пораженных тканей, хотя в последующем преобладают фиброзносклеротические изменения). Аналогично периферическую и тканевую эозинофилию выявляют преимущественно в ранней стадии заболевания, в дальнейшем она может исчезнуть или значительно уменьшиться, особенно на фоне лечения кортикостероидами.
Проведенные нами сравнительные исследования ДЭФ с системной и ограниченной склеродермией показали нозологическую самостоятельность ДЭФ, что, однако, не исключает принадлежности его к склеродермическои группе заболеваний [Гусев Н. Г. и др., 1989]. Заболевание относительно редкое, хотя это положение частично может быть обусловлено недостаточным распознаванием ДЭФ ввиду малого знакомства врачей с этой патологией.
Возрастной диапазон начала заболевания широк: от 2 до 88 лет, однако чаще ДЭФ развивается в возрасте 25—60 лет. Согласно нашим наблюдениям, средний возраст больных ДЭФ (по началу заболевания) превышает возраст больных ССД и особенно очаговой, которая нередко дебютирует в детском возрасте. В отличие от ССД мужчины заболевают ДЭФ в 2 раза чаще, чем женщины. Начало ДЭФ чаще острое, с развитием характерной симптоматики в течение первого месяца (иногда — первой недели!). Возможно и более постепенное начало с относительно медленным прогрессированием заболевания [Herson S. et al., 1984].
При осмотре выявляют двустороннее симметричное уплотнение предплечий, голеней, затем уплотнение распространяется на плечи, бедра, туловище, редко — шею и лицо. Поскольку речь идет об уплотнении фасций, прилежащих подкожной клетчатки и мышц, собственно кожа лица, шеи не изменена, что существенно отличает вид больных ДЭФ от больных ССД с характерной маскообразностью, заострением черт и т. д.
Определенное внешнее сходство с ДЭФ имеют больные с паранеопластическим псевдосклеродермическим синдромом, когда также наблюдается прогрессирующая плотность тканей с развитием контрактур, но преобладают периартикулярные изменения, индуративный процесс менее выражен и обычно имеется общая симптоматика, присущая опухолевому процессу.
В области пораженных конечностей отмечают ограничение активных и пассивных движений, тугоподвижность, стойкие сгибательные контрактуры. Артриты редки, но периартикулярные изменения в областях пораженных конечностей из-за уплотнения сухожилий, подкожной клетчатки и др. отмечаются довольно часто. Следует иметь в виду, что при паранеопластическом склеродермоподобном синдроме иногда развиваются сходные индуративные изменения конечностей с контрактурами, но отмечается преимущественное поражение периартикулярных тканей, а не фасций, как при ДЭФ.
Ранее подчеркивалось, что внутренние органы при эозинофильном фасциите в отличие от ССД не поражаются. К настоящему времени в мировой литературе имеются отдельные наблюдения ДЭФ с рестриктивным легочным фиброзом, нарушениями моторики пищевода, гепатитом и спленомегалией, перикардитом, сочетания с синдромом Шегрена, признаками дерматомиозита, которые вряд ли можно признать случайными. Альтернативной трактовкой является возможность нераспознанных атипичных форм ССД или сочетания ССД с явлениями фасциита, перекрестных или переходных клинических форм.
Из общих признаков при ДЭФ иногда отмечают небольшую лихорадку, слабость, редко похудание. Эти признаки обычно менее выражены, чем, например, при ССД и тем более — при паранеопластическом склеродермоподобном синдроме, что следует учитывать при дифференциальной диагностике с последним.
Из лабораторных сдвигов в первую очередь обращает на себя внимание гиперэозинофилия, которую можно отнести к диагностическим признакам, в значительной мере определяющим выделение ДЭФ. Гиперэозинофилия наблюдается чаще в начале заболевания, до назначения кортикостероидной терапии. Под действием кортикостероидов эозинофилия быстро снижается и исчезает; у немногих больных появляется вновь, имеет транзиторный характер.
Приблизительно в 50% наблюдений отмечено умеренное увеличение СОЭ: у отдельных больных значительное — до 83 мм/ч. У большинства больных имеется гипергаммаглобулинемия, чаще нерезко выраженная. Повышен уровень IgG и IgM, a IgA и IgE чаще в пределах нормы [Barnes L. et al., 1979].
Повышение уровня циркулирующих иммунных комплексов коррелирует с характером и активностью заболевания. Морфологическое исследование пораженных тканей, полученных методом глубокой биопсии, позволяет уточнить сущность патологического процесса и является наряду с клиническим следованием больных основой диагностики ДЭФ. Отмечена преимущественная локализация патологического процесса в фасциях прилежащих тканях. Утолщенная фасция содержит набухшие коллагеновые волокна, отечна и инфильтрирована лимфоплазмоцитарными клетками, гистоцитами, нередко эозинофилами. В биопсиях, проведенных на более поздних стадиях болезни, в фасции и прилегающих тканях отмечено преобладание склеротических процессов, уменьшение и исчезновение клеточных инфильтратов.
Эволюция и прогноз болезни чаще благоприятны, особенно фоне лечения кортикостероидами. Возможно полное выздоровление, в других наблюдениях при положительном эффекте в целом сохраняются индуративные изменения и контрактуры пальцев.
Читайте также: