
Врожденные заболевания костной системы
Генетические заболевания обусловлены патологическими нарушениями строения генома. "Дефектный" ген может быть получен от одного из родителей и проявиться как на 100%, так и на 10%. А вот болезни с наследственной предрасположенностью значительно отличаются от генетических. Если последние излечить невозможно, то заболевания, к которым человек имеет наследственную предрасположенность, возможно нивелировать рациональным питанием, здоровым образом жизни и профилактическими мерами.
Пять генетический заболеваний позвоночника и костей
Такие болезни напрямую связанны с нарушениями генома и проявляются в виде дефектов развития скелета человека. Генетические заболевания обусловлены нерациональным формообразованием ткани или нарушениями роста. Подобные болезни носят в медицине общие название - дисплазии.
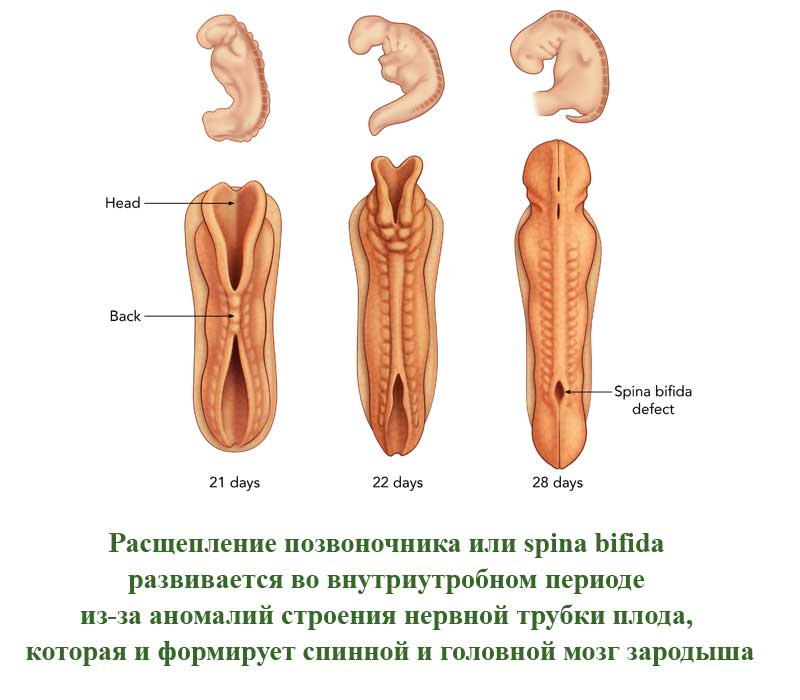
Это порок развития позвоночного столба, которое проявляется в виде недоразвитых позвонков. Такие позвонки не сомкнуты, через щель может быть виден спинной мозг. Заболевание развивается во внутриутробном периоде из-за аномалий строения нервной трубки плода, которая и формирует спинной и головной мозг зародыша. Расщепление позвоночного столба может проявляться и в закрытом виде, когда спинной мозг не виден снаружи.
В легких случаях заболевание могут обнаружить лишь при рентгеновском обследовании. А вот при самых серьезных формах болезни у ребенка могут сразу же образовываться свищи в полости позвоночника. Очень часто заболевание в тяжелых формах сопровождается параличом нижней части тела.
В более, чем 80% случаев, расщепление позвоночника сопровождается гидроцефалией спинного мозга и пороками развития головного мозга, а также - черепа.
По американской статистике, заболевание встречается у одного пациента из 1500. Российская статистика приводит следующие данные - 3 случая на 10000 человек. Однако, многие случаи расщепления позвоночника на территории СНГ остаются нераспознанными у новорожденных из-за легкой формы болезни.
Болезнь часто именуют остеопетрозом. Может протекать в двух формах:
- замедленной;
- злокачественной.
Генетическое заболевание встречается с частотой в 1 случай на 20000 пациентов. Для остеопетроза характерны такие симптомы:
- повышенная ломкость костей;
- увеличение плотности костной ткани;
- уменьшение размеров костномозговых лакун;
- нарушение гемопоэза;
- уменьшение массы костного мозга.
Генерализированный остеоклероз проявляется в достаточно раннем возрасте в виде разных беспорядочных слоев клеток костной ткани, увеличения общей массы костей и замедленном росте скелета.
При злокачественном течении болезни часто возникают внезапные переломы костей, развивается геморрагичекий синдром, жировая дистрофия органов, нарушается дентиногеез. Характерен очень небольшой рост.
В случае замедленного остеопетреза болезнь может быть выявлена лишь в 50% и протекать абсолютно бессимптомно. Выявляют заболевание случайно во время рентгена. В некоторых случаях может наблюдаться симптоматика синдрома "Кость внутри кости".
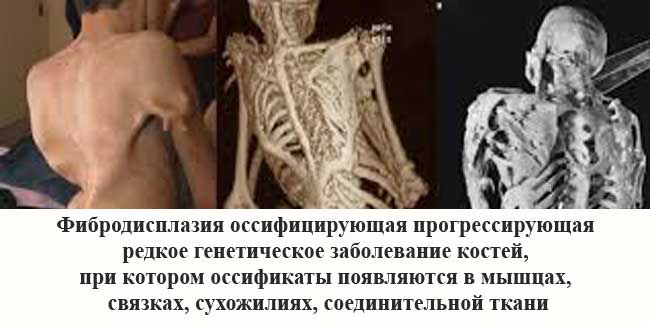
ФОП - это генетическое и очень редкое заболевание костей. При такой болезни организм начинает формировать новую костную массу в виде оссификатов в ненадлежащих местах тела, а именно внутри:
- соединительных тканей;
- связок;
- мышц;
- сухожилий.
К образованию оссификатов в организме может привести абсолютно любая травма: порез, операция, ушиб, внутримышечная инъекция или перелом. Поэтому образования такого типа удалять нельзя - на их месте костная ткань разрастется еще больше. По физиологическим признакам оссификаты совершенно не отличаются от здоровых костей.
Проблема лишь в неправильном расположении образования костной ткани. Возникает ФОП из-за мутаций гена ACVR1/ALK2. Данный ген кодирует рецептов костного морфогенетического белка. Носителем гена быть невозможно, его наличие в теле всегда вызывает развитие фибродисплазии оссифицирующей. Передается заболевание по наследству и на данный момент является неизлечимым.
Такие заболевания характеризуются чрезмерным развитием костной массы. Носят общее название - остеохондродисплазии. Гиперостозы возникают из-за генетических нарушений и патологий остеобластов и остеокластов. Наиболее часто встречаются такие формы остеохондродисплазий:
- Болезнь Лери или мелореостоз;
- пикнодизостоз.
Мелореостоз чаще всего поражает мужчин, может развиться в любом возрасте. Характеризуется болезнь избыточным образованием эндостальной или периостальной кости. Процесс может происходит в двух зонах одновременно. Зарождается болезнь Лери с поражения нижних конечностей. Процесс может переходить на все суставы, отдельные кости таза, позвоночный столб, ребра и даже череп. Все пораженные кости довольно слабо изменены и деформированы, кортикальный стой утолщен, а костномозговая полость сужена неравномерно.
Мелореостоз может протекать совершенно бессимптомно продолжительное время, однако, при значительном уменьшении габаритов костномозговых лакун развивается болевой синдром в пораженной конечности. Нога при этом может укорачиваться или увеличиваться, развивается анкилоз сустав, нарушается гемопоэз.
Пикнодизостоз проявляется в виде карликовости и остеоскрероза. В основе заболевания лежит неравномерное, чрезмерное и очаговое периостальное развитие компактной кости. Развивается явная деформация скелета в виде:
- сколиоза;
- кифоза;
- гипоплазии ключиц;
- укорочении пальцевых фаланг;
- уменьшении длины предплечий.
В молочных зубах ребенка быстро развивается кариес, склеры глаз приобретают характерных болезни голубой оттенок. На продолжительности жизни пикнодизостоз не сказывается.

Врожденная ломкость костей – это болезнь, обусловленная генетически, затрагивающая соединительную ткань, и, в частности, вызывающая нарушения строения и функции коллагена.
Как можно заключить по названию, связана с очень плохой структурой кости, которые становятся ломкими и хрупкими.
Несовершенный остеогенез – причины
Коллаген – это важный элемент соединительной ткани организма. В зависимости от того, в каком месте находится эта соединительная ткань и какие выполняет функции, соответствующий тип коллагена входит в её состав.
Несовершенный остеогенез – это заболевание, имеющее генетический фон, а мутация касается одного гена, ответственного за правильное строение коллагена – точнее, отвечающего за синтез альфа-цепи коллагена 1. Нарушения в структуре коллагена этого типа нарушают структуру и прочность костей, сухожилий, кожи, а также склер.

Заболевание наследуется чаще по аутосомно-доминантному принципу. Это означает, что наличие одного дефектного гена, полученного от одного из родителей, достаточно, чтобы появились симптомы болезни.
В зависимости от того, в какой степени происходит мутация, меняется степень тяжести сопутствующих болезни симптомов. Мутация может лишь незначительно снижать содержание коллагена в соединительной ткани, что отражается в легком течение болезни. Значительные нарушения, охватывающие процесс синтеза коллагена, могут быть причиной очень тяжелого состояния здоровья.
У некоторых людей болезнь передаётся по аутосомно-рецессивному принципу, это значит, что для того, чтобы дело дошло до появления симптомов, необходимо повреждение обоих генов, полученных от обоих родителей. Так может случиться, если каждый из родителей является носителем генетической мутации.
Симптомы врожденной хрупкости костей
Даже если заболевание обнаруживается у нескольких членов семьи, то ход его может существенно отличаться. Все случаи, однако, характеризуются различной степенью ослабления костной структуры, повышенной хрупкостью костей, склонностью к переломам. Повреждения могут возникнуть при ушибе, который у здорового человека ничего не вызовет, а в крайне тяжелых случаях повреждение костей возникают даже в состоянии покоя.
Среди других часто встречающихся симптомов:
- низкий рост
- деформации костей
- гипоплазия зубов
- голубые глаза
- глухота (во взрослом возрасте)
- дряблость суставов
- нестабильность суставов и связок
- частые синяки
Типы врожденной хрупкости костей
Существует несколько типов врожденной хрупкости кости. Самая мягкая форма – 1 тип, наиболее распространенный, характеризующийся низкой хрупкостью кости, дети не слишком малы и деформации костей не наблюдаются. Когда ребенок начинает делать первые шаги, появляются первые переломы, в том числе не только длинных костей, но и небольших костей рук или ног.
К сожалению, эта тенденция сопровождает ребёнка до достижения зрелости. В взрослом возрасте остеопоротические изменения в костях появляются намного раньше, чем у здоровых людей, и усиливается потеря слуха.
Тип 2 врожденной хрупкости костей называется летальной формой. Болезнь проявляется уже во время внутриутробной жизни, что приводит к ярко выраженным переломам и деформациям, часто заканчивается смертью плода. Рождение ребенка с врожденной хрупкостью костей типа 2 не даёт ему небольшие шансы на выживание в течение нескольких лет. Как правило, такие дети умирают в раннем возрасте.
Типы 3-4 – это умеренная и тяжелая форма, когда наблюдаются выраженные деформации костей, нарушения имеют разную форму и тяжесть. Они представляют собой промежуточные формы между слабой формой и летальной и могут давать разнообразную клиническую картину.
Повреждения костей могут появиться ещё до рождения ребенка. В более тяжелых случаях у ребёнка задерживается рост, деформируется осанка. Такие люди передвигаются на коляске, потому что болезнь делает невозможным нормальное функционирование. Гораздо раньше у таких людей появляется глухота. Больные требуют постоянных ортопедических консультаций.
Лечение врожденной хрупкости костей
Из-за генетического происхождения заболевания, не существует возможности полностью вылечить и внедрить эффективную терапию.
Лечение сводится к минимизации числа переломов, предотвращению больших деформаций и уменьшению боли. Применяется терапии бисфосфонатами, то есть препараты, используемыми в лечении остеопороза. Контролируют органы слуха, а также оценивают общее состояние больного, чтобы исключить проблемы в других системах организма.
Продолжительность жизни больных с врожденной хрупкостью костей зависит, в первую очередь, от типа заболевания. При типе 2 больной может не отличаться от общей популяции. При более тяжелых формах ожидаемая продолжительность жизни короче, чем у населения в целом, однако, это следует не из частых переломов, а из сопутствующих проблем с функционированием дыхательной или сердечно-сосудистой системы, что связано с деформацией грудной клетки.
Специалисты расходятся во мнении, в каком возрасте проводить операции. Одни предпочитают делать операцию грудничкам (в 3–6 месяцев), другие – в более поздние сроки. В любом случае лечение маленького пациента с челюстно-лицевыми аномалиями должно быть закончено к шестилетнему возрасту.
Хирургические операции делаются, как правило, в несколько этапов, число которых зависит от степени выраженности патологии. В одних случаях достаточно 2–3 вмешательств, в других требуется 7 и более. При этом полностью избежать послеоперационного рубца нельзя, несмотря на все усилия пластических хирургов.
Причины возникновения челюстно-лицевых аномалий у человека до сих пор изучены недостаточно. Уже довольно давно специалисты высказывают предположение о том, что развитие подобных заболеваний связано с курением женщины во время беременности. Этот вывод подтвержден учеными из Мичиганского университета, исследовавшими более чем 2 тыс. беременностей, завершившихся рождением детей с грубыми лицевыми аномалиями. Оказалось, что вероятность рождения ребенка с расщелинами губы и неба у курившей во время беременности женщины зависит от количества выкуриваемых ею сигарет. Если беременная выкуривает 1–10 сигарет в день, то риск возникновения врожденных дефектов челюстно-лицевой области у ее ребенка на 30% выше, чем у ребенка некурящей женщины. Если же количество выкуриваемых ежедневно сигарет превышает 21, то такая вероятность увеличивается до 70%.
Ученые полагают, что наиболее действенным способом снижения количества детей с врожденными расщелинами губы и неба является борьба с курением беременных женщин. Кроме того, по мнению специалистов, широкая и эффективная пропаганда вреда курения будущих матерей позволит реально сократить количество мертворожденных, недоношенных и маловесных детей.
Дефекты некоторых генов, влияющих на образование и развитие соединительной ткани у человека, нередко приводят к непропорциональному гигантизму. Болезнь эта была описана в 1896 г. французским педиатром А.Марфаном. При наиболее ярком проявлении этой доминантной особенности на свет появляются люди с очень длинными руками и ногами и относительно коротким туловищем. Их вытянутые пальцы напоминают лапы огромного паука. Отсюда образное название этой диспропорции – арахнодактилия (от греч. dactyl – палец и Arachna – женщина, по легенде, превращенная Афиной в паука). Люди с подобными дефектами необычайно худы, их грудная клетка бывает деформирована, хрусталик глаза смещен.
Такая аномалия носит название синдрома Марфана и считается полулетальной, поскольку связана с пороками сердца. Синдром вызван наследственным пороком развития соединительной ткани и характеризуется также поражением опорно-двигательного аппарата, глаз и внутренних органов. Первопричины таких пороков недостаточно изучены.
Нередко люди с арахнодактилией умирают от аневризмы аорты – самый крупный сосуд, выходящий из правого желудочка сердца, не выдерживает давления выбрасываемой в него крови. Люди, у которых этот синдром проявляется не со всей жестокостью, доживают до зрелых лет. По счастью синдром Марфана встречается достаточно редко. Специалисты оценивают вероятность его появления как 1/50000.
Единственная компенсация, которую люди с синдромом Марфана получают от судьбы за свой порок, – повышенное содержание адреналина в крови. Как известно, этот гормон вырабатывается надпочечниками и выбрасывается в кровяное русло в момент опасности. В результате многие параметры человеческого организма (сердцебиение, давление крови) приводятся, так сказать, в боевую готовность. Таким образом, люди с синдромом Марфана всю жизнь находятся в возбужденном состоянии: адреналин постоянно подстегивает нервную систему и делает их невероятными трудоголиками.
Синдромом Марфана страдали несколько всемирно известных личностей, отличавшихся необычайной работоспособностью. Таков был лесоруб Авраам Линкольн, который благодаря постоянному самообразованию, выдающимся способностям и, главное, потрясающему трудолюбию стал президентом США. Он обладал высоким ростом – 193 см, огромными стопами и кистями рук, маленькой грудной клеткой и длинными гибкими пальцами – типичное телосложение при синдроме Марфана.

Очень похож на Линкольна по физическому складу был сын полунищего сапожника, ставший позже великим писателем XIX в., Ганс Христиан Андерсен. Его необычайное трудолюбие проявилась еще в школе. Свои литературные произведения он переписывал до десяти раз, добиваясь в конечном счете виртуозной точности и одновременно легкости стиля.
Нервное напряжение, в котором, по-видимому, постоянно находился этот талантливый человек, порождало у него множество страхов. Он боялся заболеть холерой, пострадать от пожара, попасть в аварию, потерять важные документы, принять не ту дозу лекарства.

История знает случай, когда длинные, тонкие пальцы человека с синдромом Марфана вместе с впечатляющей работоспособностью помогли их обладателю сделать фантастическую карьеру. Речь идет о знаменитом скрипаче Никколо Паганини. Гете и Бальзак так описывают его внешность в своих воспоминаниях: мертвенно-бледное, как будто вылепленное из воска лицо, глубоко запавшие глаза, худоба, угловатые движения и, самое главное, тонкие сверхгибкие пальцы, какой-то невероятной длины, как будто вдвое длиннее, чем у обычных людей. Эта чисто морфологическая особенность позволяла ему творить со скрипкой настоящие чудеса.
В толпе, слушавшей импровизации Паганини на римских улицах, одни говорили, что он в сговоре с дьяволом, другие – что его искусство является музыкой небес, в которой звучат ангельские голоса. Он играл так, что слушателям казалось, будто где-то спрятана вторая скрипка, играющая одновременно с первой. Многие вплоть до XX в. верили слухам, что в молодости Никколо прибег к помощи хирурга, который сделал ему операцию, чтобы повысить гибкость рук. Теперь-то мы знаем, что, скорее всего, своим данными он был обязан редкому генетическому отклонению.
Надо заметить, что сам по себе синдром Марфана не располагает к музыкальной одаренности. За исключением Паганини, среди больных с этим синдромом не было выдающихся музыкантов. Что же касается Паганини, то болезнь лишь придала ему большие технические возможности, а великим музыкантом, с огромным творческим наследием, включающим, кроме произведений для скрипки с другими инструментами и оркестром также более 200 пьес для гитары, он стал благодаря своему великому таланту и трудолюбию, тоже косвенно связанному с синдромом Марфана.
Из наших современников синдромом Марфана, возможно, страдал биолог Г.В. Никольский. Ко времени окончания Московского университета он имел уже пять печатных трудов. За 30 последующих лет работы число его печатных публикаций превысило 300, причем среди них было около десяти книг. Такой потрясающей работоспособностью может похвастаться далеко не каждый даже очень способный ученый! Можно ли после этого утверждать, что любые обусловленные генами нарушения в развития являются безусловно вредными?
Мышечная система является самой крупной системой органов в теле человека. Еще в древности люди подметили, что перекатывающиеся упругие мышцы похожи на шныряющих под кожей мышек, поэтому они назвали такие образования мускулами (от лат. musculus – мышка). Мышца похожа на сплетенный из волокон канат. Сверху она покрыта защитной оболочкой из соединительной ткани. Каждое волокно мышцы состоит из тончайших белковых нитей – миофибрилл (от греч. myos – мышь, мышца и лат. fibra – волокно).
Основой миофибрилл являются два белка – актин и миозин. Каждая миофибрилла состоит примерно из 2,5 тыс. белковых нитей актина и миозина. Во время сокращения мышцы они не укорачиваются, а лишь скользят друг по другу. В результате длина мышцы становится меньше, она сокращается. Представить, как происходит такой процесс, легко, если вставить пальцы одной руки между пальцами другой, держа ладони в одной плоскости.
Мышца сокращается только в том случае, если получает от нервной системы слабые сигналы – электрические импульсы. Роль нервов в сокращении мышц была впервые замечена еще древнеримским врачом Клавдием Галеном, который изучал анатомию в школах гладиаторов. Там-то он и подметил, что повреждение нервов часто приводит к потере мышечной подвижности.
Время подтвердило этот вывод. Теперь мы знаем, что для сокращения мышечное волокно оно должно получить нервный сигнал. В результате внутри волокна из специальных мембранных емкостей высвобождаются ионы кальция, которые стимулируют актин и миозин ко взаимному скольжению. Роль электрохимической изоляции мышечного волокна играет специальная оболочка – сарколемма. Запомните этот термин. Он нам еще пригодится!
У каждого человека ровно столько мышц, сколько и у Арнольда Шварценеггера, различается лишь их сила. Она зависит от числа мышечных волокон, входящих в состав мышцы, и от интенсивности приходящих к ним нервных сигналов. Такие сигналы поступают к мышцам всегда, даже во время сна. В результате каждая наша мышца постоянно находится в состоянии тонуса (от греч. tonos – напряжение), т.е. слегка напряжена. Тонус мышц исчезает только после кончины человека.
Школьные знания из области анатомии и физиологии мышц позволяют сделать следующий вывод. Врожденные дефекты мышечной системы могут возникать как в результате дефектов мышечных белков, так и вследствие нарушений иннервации разных групп мышц. Нервно-мышечные заболевания, следствием которых является их быстрая утомляемость, слабость, снижение мышечного тонуса и даже атрофия, называются миопатиями. Рассмотрим некоторые врожденные формы таких недугов.
Наиболее распространенным наследственным нервно-мышечным заболеванием человека является мышечная дистрофия Дюшенна. Ее частота среди новорожденных мальчиков составляет около 1/5000.
Причиной дистрофии этого типа являются мутации в одном единственном гене. Он хранит информацию о строении белка, весьма образно названного дистрофином. Этот белок входит в состав сарколеммы (оболочки) мышечных волокон, обеспечивая стабильность этой своеобразной изолирующей упаковки мышц. Дефектный белок не в состоянии выполнять эту функцию, следствием чего является нарушение целостности мембраны. Это приводит к дегенерации мышечных волокон.
Мембраны мышц становятся проницаемыми, словно дырявый полиэтиленовый пакет. Это приводит к оттоку ферментов из мышц в сыворотку крови. На начальной стадии заболевания дегенерация мышечных волокон еще компенсируется активной регенерацией мышечных фибрилл благодаря делению и слиянию вспомогательных клеток. Однако с возрастом компенсация становится все менее эффективной, мышечная слабость прогрессирует. Мышцы постепенно замещаются фиброзной и жировой тканью. Мальчики к 12 годам уже оказываются прикованы к креслу-каталке. Смерть при мышечной дистрофии Дюшенна обычно наступает в возрасте около 30 лет в результате нарушения работы сердца и диафрагмы.
Ген дистрофина является самым большим из известных генов человека и составляет почти 0,1% всей его ДНК. Он находится в Х-хромосоме. В результате наследование дистрофии Дюшенна сцеплено с полом. Страдают от этого недуга в основном мальчики. Ген дистрофина работает не только в клетках мышц, но и во многих тканях, в головном мозге, в сетчатке и в клетках потовых и слюнных желез. Однако в первую очередь дефект в его работе проявляется именно в мышцах.
Менее злокачественно развивается миодистрофия Беккера. Причина ее та же – дефект белка дистрофина. Однако при миодистрофии Беккера, в отличие от миодистрофии Дюшенна, этот белок все же продолжает работать, хотя и хуже, чем в норме.
Миодистрофия Беккера развивается медленно, особенно у низкорослых детей. Многие годы они сохраняют удовлетворительное физическое состояние, и только сопутствующие заболевания и травмы приковывают их к инвалидной коляске. Дистрофия Беккера менее распространена, чем миодистрофия Дюшенна.
Помимо миодистрофий Дюшенна и Беккера существует еще несколько форм врожденных миопатий. Например, юношеская форма миопатии Эрба–Рота возникает в возрасте
10–20 лет, когда начинается атрофия мышц плечевого пояса и рук, а затем — тазового пояса и ног. Во время ходьбы больной переваливается с выпяченным вперед животом и отодвинутой назад грудной клеткой. Чтобы встать из положения лежа, ему надо повернуться на бок и, опираясь руками на бедра, постепенно поднять свое туловище. Болезнь со временем медленно прогрессирует.
Существует плече-лопаточно-лицевая форма миодистрофии (Ландузи-Дежерина), которая может начаться в возрасте от 6 до 52 лет (чаще в 10–15 лет). Для нее характерны поражение мышц лица и постепенная атрофия мышц плечевого пояса, туловища и конечностей. На ранних стадиях болезни веки плохо смыкаются, не закрываются полностью. Губы также не смыкаются, что создает проблемы с дикцией и невозможность надуть щеки. Заболевание протекает медленно. Долгое время больной может передвигаться и сохранять трудоспособность, а затем через 15–25 лет постепенно начинают атрофироваться мышцы тазового пояса и ног, что затрудняет передвижение.
При невральной амиотрофии Шарко–Мари происходит постепенная атрофия мелких мышц стоп, затем атрофируются мышцы голеней и нижней части бедер. Мышцы средней и верхней частей бедер при этом не изменяются, и бедро приобретает форму бутылки, опрокинутой горлышком вниз. Затем постепенно атрофируются мышцы кистей рук и предплечий. Мышцы туловища, плечевого пояса и лица не поражаются. Заболевание возникает в возрасте 18–25 лет, медленно прогрессирует и стабилизируется. В его основе лежит нарушение иннервации соответствующих групп мышц.
Снижение тонуса мышц называют амиотонией. При врожденной амиотонии Оппенгейма мышцы новорожденного недоразвиты, их дистрофия является вторичной. У новорожденных болезнь не прогрессирует, но респираторные инфекции могут вызвать в этом случае серьезное воспаление, которое нередко приводит к смерти на первом году жизни. С возрастом двигательная функция мышц при амиотонии Оппенгейма улучшается.
Лечение мышечных дистрофий направлено на замедление дистрофических процессов в мышцах, а при возможности – на их полное прекращение. К сожалению, радикального метода лечения миодистрофий пока нет. Некоторые надежды вселяет генная терапия, которая начинает медленно внедряться в медицинскую практику. Для лечения миодистрофий применяют витамин В1, внутримышечные инъекции аденозинтрифосфата (АТФ), делают переливание крови. Из народных средств применяют проросшие зерна пшеницы, ржи, пчелиное маточное молочко, траву спорыша, хвоща полевого, льнянку обыкновенную, женьшень, корневища топинамбура.
Известно, что врожденные мышечные дистрофии бывают не только у людей. Например, собаки породы золотистый ретривер часто являются носителями дефектного гена белка дистрофина. При этом у них развиваются типичные клинические проявления дистрофии Дюшенна. Таких собак экспериментаторы давно используют в качестве модельных объектов для изучения особенностей течения данного заболевания и поисков способов его лечения. Аналогичная ситуация с дефектным геном дистрофина возникает и у некоторых кошек. Нередко эти несчастные животные погибают в результате нарушений работы мышц диафрагмы. Генетики вывели даже особую линию лабораторных мышей с точечной мутацией в гене дистрофина. Оказывается, таким мышам можно помочь, вводя в их мышцы здоровые эмбриональные мышечные клетки.
Аналогичные эксперименты проводят и на больных людях. Суть метода заключается в получении мышечных клеток от здорового донора. Затем их подращивают вне организма (in vitro) и вводят в мышцы больного. Стоимость такой операции составляет около 150 тыс. долларов США. Такие опыты были проведены в шести независимых исследовательских лабораториях, а результаты доложены в Париже в 1999 г. К сожалению, ведущие авторитеты в области биологии мышц и миодистрофий на данном этапе считают этот метод абсолютно неэффективным.
Другой возможный путь – пересадка собственных стволовых клеток, полученных из костного мозга или скелетных мышц больного. При этом часть инъецированных клеток мигрирует и в скелетные мышцы, где сливается с миофибриллами, восстанавливая синтез дистрофина. Генотерапия же в данном случае пока почти бессильна. Несмотря на то что ученые умеют выделять ген дистрофина, им пока не удается доставить его по назначению, т.е. ввести в мышечные клетки, где работает его дефектная копия.
Однако ученые не опускают руки. В нашей стране исследования по генной терапии миодистрофии Дюшенна ведутся в Институте акушерства и гинекологии им. Д.О. Отта (Санкт-Петербург) в тесном контакте с ведущими лабораториями по данной проблеме в Великобритании и Италии, а также в комплексе с другими научно-исследовательскими институтами России, в частности Институтом молекулярной биологии им. В.А. Энгельгардта, Институтом цитологии в Санкт-Петербурге, Научным центром медицинской генетики и Институтом экспериментальной медицины в Санкт-Петербурге. Остается только надеяться, что в обозримом будущем эти исследования увенчаются практическим успехом.
Публикация статьи произведена при поддержке ООО "ЭЙДОС", предлагающего гибридные медицинские симуляторы для моделирования различных ситуаций – от работы бригады скорой помощи в реанимобиле, до операционных тренажеров, позволяющих провести на виртуальном симуляторе репетиции эндохирургических операций, гистероскопии и других операционных вмешательств, а также акушерских манипуляций. Широкий выбор роботов-симуляторов позволяет охватить большой спектр учебных ситуаций для подготовки врачей и медперсонала.

Костная система подвержена повреждениям, износу, инфекции, опухоли и метаболическим заболеваниям, которые приводят к повреждению костей
Скелет человека состоит из 206 костей, которые соединены вместе посредством связок и соединительной ткани. Скелет не только обеспечивает двигательную функцию, но и защищает жизненно важные органы (мозг, сердце, легкие и органы брюшной полости). Однако наша костная система подвержена повреждениям, износу, инфекции, опухоли и метаболическим заболеваниям, которые приводят к повреждению костей, что может стать опасным для жизни. Ниже приведены некоторые общие заболевания костной системы.
Общие заболевания костной системы
Артрит существует в двух основных формах. Артроз – это износ наших костей и суставов, который происходит с возрастом. Ожирение является одним из важных факторов, которые могут ускорить остеоартрит, особенно коленей и бедер. Все стыки костей выстланы хрящом и синовиальной жидкостью, которые помогают смазывать сустав во время движений. Со временем эти ткани разрушаются и стираются, что приводит к формированию костной шпоры, совместному сужению, воспалению и боли. Лечение тяжелого остеоартрита заключается в применении обезболивающих средств, а также инъекций стероидов. В запущенных случаях требуется замена сустава.
Аутоиммунный артрит возникает, когда организм атакует свои суставы и повреждает их. Ревматоидный артрит является одним из примеров таких заболеваний. Со временем они приводят к уничтожению суставов и хронической слабости. Лечение направлено на управление болью и модулирует иммунную систему, что позволяет ограничить ее дальнейшее разрушение.
Остеопороз: заболевание костной системы, характеризующееся снижением плотности костей
Остеопороз – это уменьшение прочности и минеральной плотности костей. Возраст, гормональный статус и диета играют жизненно важную роль в развитии остеопороза. Кости становятся постепенно слабыми и склонны к переломам с незначительными травмами.
Рахит: заболевание костной системы, связанное с дефицитом витамина Д
Рахит/остеомаляция возникает вследствие сильного дефицита кальция, витамина Д и фосфатов. Кости размягчаются и становятся слабыми, теряют свою нормальную форму. Отмечаются боль в костях, судороги и скелетные деформации.
Бурсит: заболевание костной системы, связанное со скоплением жидкости вокруг суставов
Раковые заболевания костной системы
Белые клетки крови – лейкоциты – частично вырабатываются в костном мозге. Как правило, целый ряд видов рака крови называют лейкозом. Начало лейкоза вообще является коварным. Пока не образовалась критическая масса атипичных клеток, у большинства людей это заболевание протекает бессимптомно. Ранние симптомы лейкоза: костные боли, сильная усталость, ночные поты, необъяснимая потеря массы тела и кровоточивость десен.
В кости также могут возникать раковые опухоли. Рак костей может быть основным типом рака, а также может появиться вследствие метастазирования расположенной в другом месте раковой опухоли (легких, молочной железы и простаты). Основные типы рака кости относят к остеосаркоме и саркоме Юинга.
Врожденные заболевания костной системы
Косолапость – это врожденный дефект в развитии одной или обеих ног, которые выгнуты внутрь и вниз. В результате этого заболевания ребенку очень сложно научиться ходить. Часто необходима специализированная ортопедическая терапия или операция.
Спина бифида – это врожденный дефект, который связан с неполным закрытием позвонка вокруг позвоночного канала. Многие люди имеют слабые формы этого заболевания и даже не знают об этом. Более тяжелые формы заболевания сопровождаются нервными дефектами, трудностями при ходьбе, а также проблемами с функцией кишечника и мочевого пузыря.
Другие заболевания костной системы
Несовершенный остеогенез – это спектр заболеваний костной системы, начиная от легких до тяжелых и опасных для жизни. Люди с этими заболеваниями склонны к переломам даже при незначительных травмах. Наиболее тяжелые формы этих заболеваний приводят еще к внутриутробной смерти. У людей с этими заболеваниями склеры (белая часть глаза) часто имеют голубоватый оттенок.
Остеопетроз (мраморная болезнь) – редкое заболевание костной системы, при котором кости становятся буквально окаменевшими и могут легко ломаться.
Болезнь Педжета приводит к тому, что кости ломаются быстрее, чем они могут восстанавливаться. Обычно в организме этот процесс находится в балансе. Однако, при болезни Педжета происходит ускоренный распад костной ткани, а кости становятся хрупкими. Это приводит к повышенному риску переломов.
Таким образом, заболевания костной системы можно отнести к 4 основным группам: 1) врожденные/генетические (косолапость, спина бифида, остеопетроз, болезнь Педжета; 2) возрастные (остеопороз, артрит, артроз); 3) раковые (рак кости и лейкоз); 4) обусловленные травмой (тендинит, переломы).
Читайте также: